分子动力学NAMD部署文档 |
您所在的位置:网站首页 › archlinux官方文档 › 分子动力学NAMD部署文档 |
分子动力学NAMD部署文档
|
高性能计算实战——NAMD
概述
NAMD(NAnoscale Molecular Dynamics)是用于在大规模并行计算机上快速模拟大分子体系的并行分子动力学代码。目前 NAMD 还支持在 GPU 加速器上的运算。NAMD 具有非常强的大规模并行计算能力,已经实现了在上千个处理器上的并行计算,对包含超过三十万个原子的大分子系统进行模拟。 软件环境 Fortran90、gcc编译器 单精度FFTW NAMD 其他:CUDA等 FFTWFFTW ( the Faster Fourier Transform in the West) 是一个快速计算离散傅里叶变换的标准C语言程序集,其由MIT的M.Frigo 和S. Johnson 开发。可计算一维或多维实和复数据以及任意规模的离散傅里叶变换。FFTW 受到越来越多的科学研究和工程计算工作者的普遍青睐,并为量子物理、光谱分析、音视频流信号处理、石油勘探、地震预报、天气预报、概率论、编码理论、医学断层诊断等领域提供切实可行的大规模 FFT 计算。 CUDA至官方网站下载对应操作系统的安装包。可参阅其中安装手册下载,安装完成后配置环境变量例如: export PATH=$DIR/cuda/bin:$PATH export LD_LIBRARY_PATH=$DIR/cuda/lib64:$LD_LIBRARY_PATH export C_INCLUDE_PATH=$DIR/cuda/include:$C_INCLUDE_PATH 安装步骤 NAMD在 NAMD 官网下载合适版本,以源码为例,先解压缩 tar xzvf NAMD_2.13_Source.tar.gz其中包含charm安装包,继续解压缩 cd NAMD_2.13_Source tar xvf charm-6.8.2.tar 依赖库安装主要为tcl与FFTW,首先获取安装包 wget http://www.ks.uiuc.edu/Research/namd/libraries/fftw-linux-x86_64.tar.gz wget http://www.ks.uiuc.edu/Research/namd/libraries/tcl8.5.9-linux-x86_64.tar.gz wget http://www.ks.uiuc.edu/Research/namd/libraries/tcl8.5.9-linux-x86_64-threaded.tar.gz可能实际下载时平台的wget指令无法使用,此时需要先手动下载文件至本地再传输至高性能计算服务器上。 Windows环境下可以使用WinSCP软件,WinSCP是一个Windows环境下的开源图形化SFTP客户端。同时支持SCP协议。它的主要功能就是在本地与远程计算机间安全的复制文件。 打开软件后输入主机地址,用户名和密码即可连接 详细安装和使用方法可以参考官方网站 进行安装 tar xzf fftw-linux-x86_64.tar.gz mv linux-x86_64 fftw tar xzf tcl*-linux-x86_64.tar.gz tar xzf tcl*-linux-x86_64-threaded.tar.gz mv tcl*-linux-x86_64 tcl mv tcl*-linux-x86_64-threaded tcl-threaded 执行编译 编译charm++并行库根据需求选择不同模式 tar xvf charm-6.8.2.tar cd charm-6.8.2/ ./build charm++ verbs-linux-x86_64 gcc smp --with-production #分布式计算模式 ./build charm++ multicore-linux64 gcc --with-production #单节点 multicore 模式 编译NAMD主程序编译单节点模式: ./config Linux-x86_64-g++ --charm-arch multicore-linux64-gcc --with-cuda --cuda-prefix /usr/local/cuda cd Linux-x86_64-g++/ make编译多节点并行模式: ./config Linux-x86_64-g++ --charm-arch verbs-linux-x86_64-smp-gcc --with-cuda --cuda prefix /usr/local/cuda cd Linux-x86_64-g++/ make 编译完成后会生成 charmrun,namd2 等文件。至此NAMD已经安装完毕 VMDVMD 是一个分子可视化程序,该程序采用3D图形以及内置脚本来对大型生物分子系统进行显示、制成动画以及分析等操作。 测试环境时安装的版本为vmd-1.9.2,在官方网站下载安装包后,上传至高性能计算服务器。 首先解压缩并进入目录 tar xzvf vmd-1.9.2.bin.tar.gz cd vmd-1.9.2首先修改configure文件中安装路径,即install_bin_dir和install_library_dir两个变量 vim configure执行configure操作,对于不同体系结构的高性能计算机,详细的配置选项可在[官网][https://www.ks.uiuc.edu/Research/vmd/doxygen/configure.html]查看 ./configure之后进入src目录下安装 cd src make install安装成功后,运行vmd可以看见相关信息
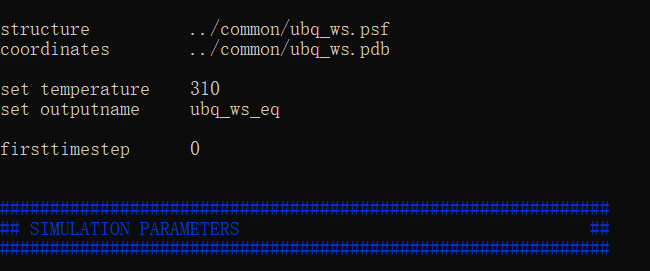
主要包括生成结构文件、溶质化、球状水体泛素的分子动力学模拟、立方体水泛素的分子动力学模拟等一系列步骤。 基本准备首先需要在官网下载运行所需要的一个数据包。 分子动力学模拟需要准备各种数据文件包括 分子的PDB文件。存储分子所有原子的坐标。 分子的PSF文件。存储分子的结构信息。 力场参数文件。 模拟的配置文件。创建PSF文件 vim ubq.pgn输入如下内容 package require psfgen topology top_all27_prot_lipid.inp pdbalias residue HIS HSE pdbalias atom ILE CD1 CD segment U {pdb ubqp.pdb} coordpdb ubqp.pdb U guesscoord writepdb ubq.pdb writepsf ubq.psf配置文件 创建配置文件,修改部分参数如图中列举的就有 psf和pdb文件的位置 环境温度temperature 定义变量outputname并赋值为ubq_ws_eq 设置起始时刻这里只截取了部分,实际还有许多参数,详细配置可以查看官方手册。
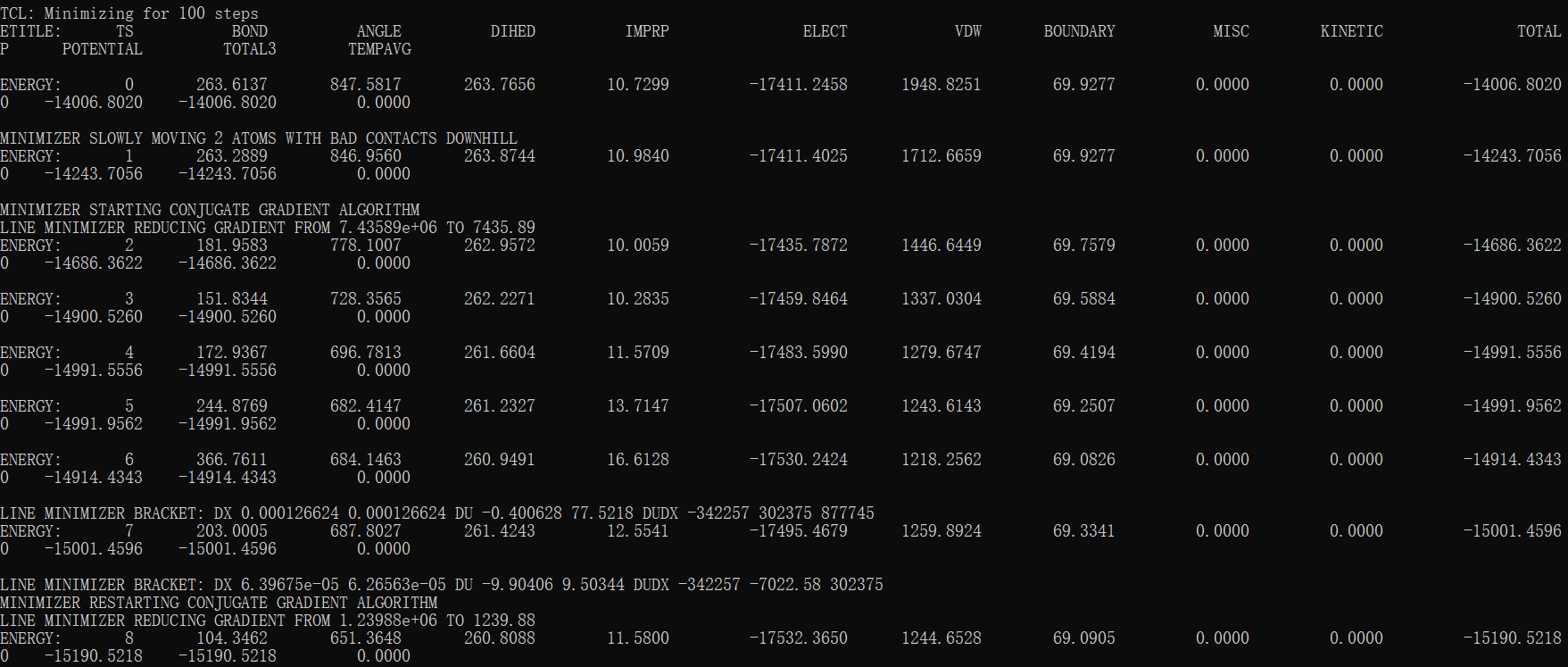
模拟计算 ./namd2 ubq_ws_eq.conf > ubq_ws_eq.log 实验结果编写slurm脚本 #!/bin/bash #SBATCH -J namd #SBATCH -p cpu-low #SBATCH -N 4 #SBATCH -n 8 #SBATCH -t 15:00 #SBATCH -o out #SBATCH -e err ./namd2 +p8 +setcpuaffinity namd-tutorial-files/1-2-sphere/ubq_ws_eq.conf > test_4_8_log执行结果后,可以查看输出文件,得到每一步的模拟信息。
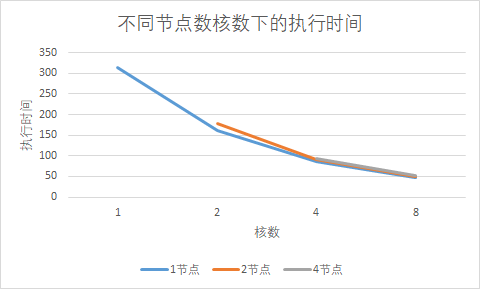
可以在运行时指定分配的节点数、核数,在运行时会输出相关配置信息,在计算结束后查看对应结果。 每次选择不同的节点数核数,计算后查看cpu时间,每组环境下进行多次测试,并观察其变化趋势
可以发现在使用多核并行的情况下有较好的加速比。 导入数据计算假设新的数据已经有以下文件,并需要根据特定需求计算 蛋白质分子的PDB文件。存储蛋白质分子所有原子的坐标。 蛋白质分子的PSF文件。存储蛋白质分子的结构信息。 力场参数文件。运行前,需要编写配置文件,主要目的在于 指定输入数据的路径 设置参数 修改计算相关配置将输入数据都放在上一级目录的common文件夹中,即../common,则可以这样编写 |
【本文地址】
今日新闻 |
推荐新闻 |