如何快速批量找到转录本序列的开放阅读框(ORF) |
您所在的位置:网站首页 › Genbank怎么看自己上传上去的序列 › 如何快速批量找到转录本序列的开放阅读框(ORF) |
如何快速批量找到转录本序列的开放阅读框(ORF)
|
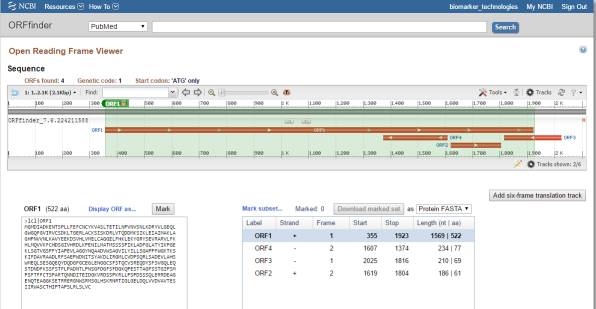
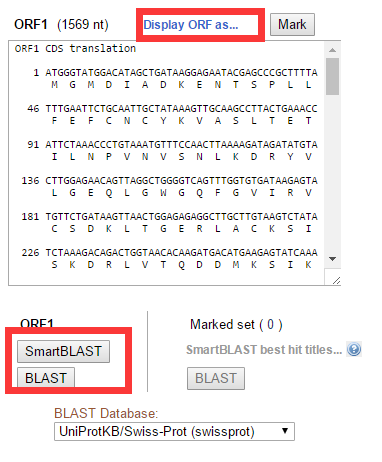
点击不同的ORF即可给出相应的氨基酸序列,也可以设置为显示DNA序列或者DNA和protein相互比较的序列形式,如下图标红框的部位。接下来的问题是,如何知道预测出来的氨基酸序列是否可靠呢?一般最长的氨基酸序列就是比较可靠了,也可以直接点击BLAST对得到的氨基酸序列进行blastp分析,看能否比对到已知的蛋白序列上。
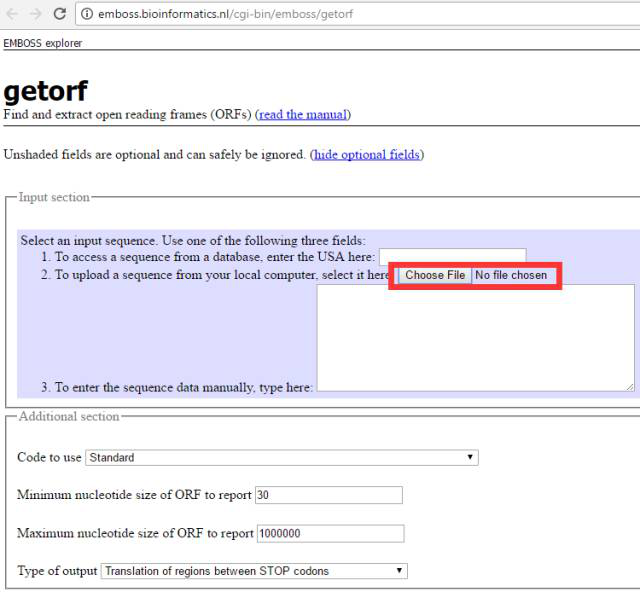
☼ 点评 NCBI的ORF Finder简单易用,结果的展现形式也很直观,还能直接对得到的氨基酸序列进行blastp分析,以验证ORF的可靠性。但缺点是不能进行批量处理,只能一个一个转录本序列进行分析,这对于手上握有大量数据的同学,就有点慢了,下面介绍一款可以批量进行ORF预测的软件。 2.getORF getORF也是一款经典的用于分析转录本序列ORF的软件,属于EMBOSS软件包。百度搜索一下getORF,会出现多个条目,是不同的科研单位搭建的在线运行getORF的平台,都可以用,小编在这里推荐一个速度比较快的平台http://emboss.bioinformatics.nl/cgi-bin/emboss/getorf,界面如下:
getORF与ORF Finder最大的不同就是可以上传fasta格式存储的序列(不知道什么是fasta格式的同学请自行百度),这样就可以一次上传很多条转录本序列,批量进行处理。其它的设置与ORF Finder相同,可以设置ORF长度最大和最小的限制,也可以选择是否以ATG开头(Translation of regions between START and STOP codons),默认是不以ATG开头(Translation of regions between STOP codons)。
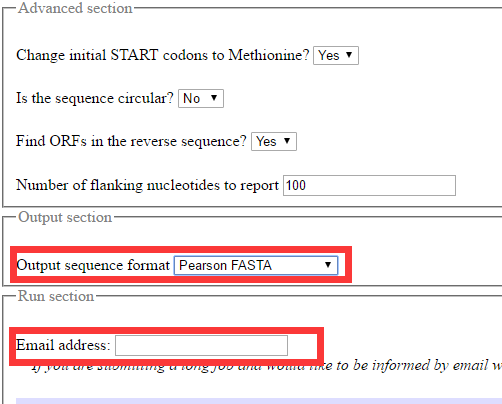
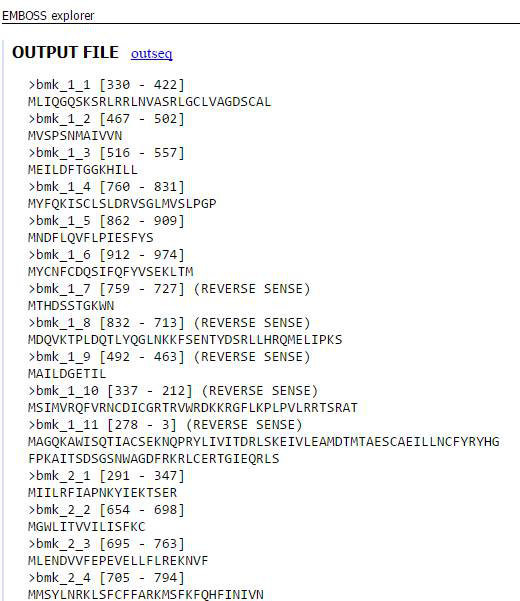
在高级设置里面还可以有多种设置,默认即可。输出的格式也可以有多种选择,还可以填写自己的email地址,会发送结果文件的链接到邮箱。最终的输出结果如下图,同一个转录本的不同ORF用后缀的数字表示(_1,_2…)。输出结果可以进行复制保存,然后提交到blastp进行分析。
☼ 点评 getORF最大的优势是可以进行批量处理,预测得到的ORF也很多,但界面的呈现不如ORF Finder直观。返回搜狐,查看更多 |
【本文地址】
今日新闻 |
推荐新闻 |