利用GEM |
您所在的位置:网站首页 › GEM软件 › 利用GEM |
利用GEM

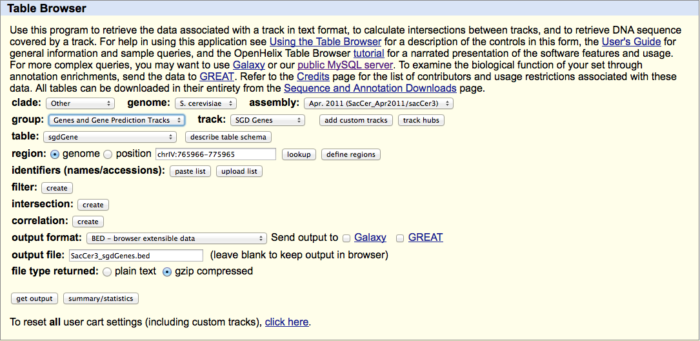
现在测序应用越来越广泛,各种Seq满天飞,譬如ChIP-Seq, DNase-Seq等等。在align完测序的reads后,有些位置reads多有些位置reads 少。除了本身的生物特性外是否还与其他的因素有关呢? 当然最主要的因素应当是基因组序列本身,毕竟基因组不是简单的ACGT的完全随机。另外一个因素就是reads的长度。今天给大家介绍利用GEM-library(论文Derrien et al in 2012 [1],软件下载 [2])创建基因组Mappability文件。这个就是基于基因组本身创建出在特定条件下(譬如reads长度50bp,允许2个mismatch)基因组各位置unique reads的覆盖情况。 下面主要利用https://wiki.bits.vib.be/index.php/Create_a_mappability_track#cite_note-2 网站的基本方法来给大家介绍一下如何以酵母基因sacCer3为例生成Mappability文件。 1.下载参考数据 1.1 Download the yeast reference genome data from the UCSC table repository UCSC data can be obtained from their FTP server http://hgdownload.soe.ucsc.edu/goldenPath/sacCer3[3]other files are built from the table browser with interaction in your browserWe use below Kent tools (unix version from [4], also available for macOSX[5]) used by the UCSC team to produce the Genome browser system. Some of these tools should be present on your machine in order to repeat the code below. # create a folder to store all results basefolder="~/Desktop/alignability/sacCer3_alignability" mkdir –p ${basefolder} && cd ${basefolder} # download the 2bit file wget http://hgdownload.soe.ucsc.edu/goldenPath/sacCer3/bigZips/sacCer3.2bit # extract the fasta data with Kent\'s \'twoBitToFa\' twoBitToFa sacCer3.2bit SacCer3.fa # also create a two-column list of chromosomes names and sizes with Kent\'s \'faSize\' faSize sacCer3.fa -detailed > sacCer3.sizes1.2 download additional SacCer3 annotations Get the SGD gene table as well as the GC distribution across the whole genome from the UCSC-table (http://genome.ucsc.edu/cgi-bin/hgTables[6]). 注意:the chromosome names should be identical between the 2bit-fasta and the gene table in order for IGV to display them together # create a folder to store all results basefolder="~/Desktop/alignability/sacCer3_alignability" mkdir –p ${basefolder} && cd ${basefolder} # get the sgdGene table for other purposes (this is not a BED file) wget http://hgdownload.soe.ucsc.edu/goldenPath/sacCer3/database/sgdGene.txt.gz # also get it in the BED-format \' SacCer3_sgdGenes.bed\' using the Table browser (see screen-shot below) # get and adapt GC% data in variable steps wget http://hgdownload.soe.ucsc.edu/goldenPath/sacCer3/gc5Base/sacCer3.gc5Base.varStep.txt.gz # the file: sacCer3.gc5Base.varStep.txt.gz is the raw data used to encode # the gc5Base track on sacCer3. This file was produced from the sacCer3.2bit # sequence with the kent source tree utility hgGcPercent (see below)
1.3 create a 5base step file from the variable step with Kent\'s hgGcPercent We can produce the GC% wig file directly from the 2bit genome and name it slightly differently to the downloaded version. # create a folder to store all results basefolder="~/Desktop/alignability/sacCer3_alignability" mkdir –p ${basefolder} && cd ${basefolder} hgGcPercent -wigOut \ -doGaps \ -file=stdout \ -win=5 \ sacCer3 \ sacCer3.2bit | \ gzip > sacCer3.gc5Base.varStep.wig.gz1.4 create a bigwig version for rapid visualization The wigToBigWig command is somehow sensitive to complex fasta headers. If you Fasta sequences have long names including spaces and \':\', you should better simplify it and keep only one word like \'1\' or \'chr1\' to avoid problems at this stage # create a folder to store all results basefolder="~/Desktop/alignability/sacCer3_alignability" mkdir –p ${basefolder} && cd ${basefolder} wigToBigWig |
【本文地址】