对肿瘤驱动基因及FGFR靶点的一些梳理及看法 所谓“深入方可浅出”。在分析某一个靶点之前,我们会先来分别讨论癌症驱动基因、受体酪氨酸激酶的概念,以及FGFR抑制剂开发... |
您所在的位置:网站首页 › FGFR2基因检测哪里可以做 › 对肿瘤驱动基因及FGFR靶点的一些梳理及看法 所谓“深入方可浅出”。在分析某一个靶点之前,我们会先来分别讨论癌症驱动基因、受体酪氨酸激酶的概念,以及FGFR抑制剂开发... |
对肿瘤驱动基因及FGFR靶点的一些梳理及看法 所谓“深入方可浅出”。在分析某一个靶点之前,我们会先来分别讨论癌症驱动基因、受体酪氨酸激酶的概念,以及FGFR抑制剂开发...
|
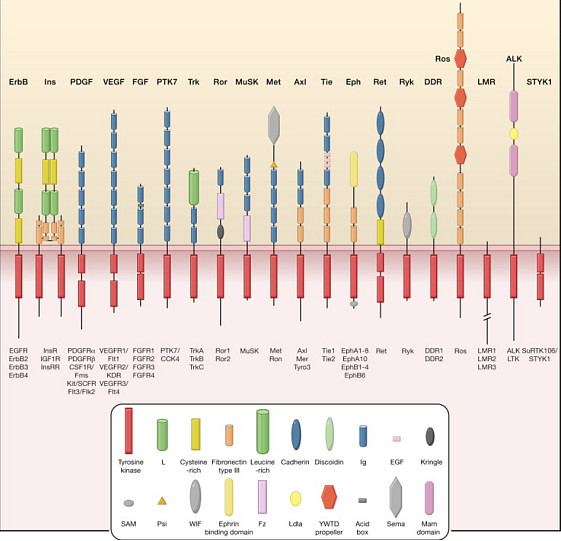
来源:雪球App,作者: 青侨阳光,(https://xueqiu.com/3770558188/160191113) 所谓“深入方可浅出”。在分析某一个靶点之前,我们会先来分别讨论癌症驱动基因、受体酪氨酸激酶的概念,以及FGFR抑制剂开发现状。这些讨论提供了比较简洁但可能有用的行业背景图景,为我们分析产品的竞争力提供重要的参考基础。 一、肿瘤突变与驱动基因 肿瘤的本质特征,某种程度上就是一系列突变累积后产生的生长失控。 人体是个非常精密的系统,一个细胞要想累积突变成为可迁移、能复制、不被逮捕击杀的恶性癌细胞,是需要突破人体内的重重防线,包括躲避巨噬细胞和T细胞的“捕杀”、抑制细胞凋亡和自噬的“监控”、修改细胞周期的调节、适应缺氧等营养环境等重重困境,同时需要获得细胞迁移、血管新生补给、快速分裂等等技能;要“集齐癌变所需的各种技能”,绝非易事。 我们可以先看一组数据:人体有40-60万亿个细胞,每个细胞有30亿个碱基,即使是人体高保真的DNA聚合酶的错配率低至10-9-10-10,即使假设人体细胞平均1-3年才更新一次,也意味着每个人平均每天都“创造”了数千亿次的碱基突变!当然,这些突变绝大部分会是无义而离散的改变,只有极少数突变会对细胞的遗传环境产生显著的影响。 这些改变中,隐藏了未来细胞癌变的基础;只不过绝大部分“有癌变潜力的细胞”都会被人体的复杂监控系统“扼杀”在摇篮中(比如被巨噬细胞吞噬、被T细胞杀伤、或者诱发细胞凋亡和自噬等)。在这些有潜力癌变的细胞中,又只有极少数最终能“积累足够的突变”完成真正的恶性癌变。所以,细胞癌变的过程,某种程度上也是累积突变的过程;到肿瘤晚期,每个癌细胞的突变密度是非常高的。这也是肿瘤高异质性、很容易治疗耐药进展的重要原因。 但不同的突变,在临床上的意义上是不同的。某些关键基因的关键突变,会是驱动肿瘤发生和发展的关键因素,这些关键基因突变后翻译产生的异常蛋白,会继发激活更高频率更大范围的基因突变,从而推动肿瘤进程。有时候也会发生上游的某些突变或改变,导致某个关键基因过度表达或过度激活,也会推动肿瘤进程。这些异常突变或异常激活的关键基因,被称为肿瘤的驱动基因(cancer driver gene)。 肿瘤驱动基因的概念,实际上为肿瘤治疗提供了一种理论基础:如果我们能够找出某个肿瘤的驱动基因,如果我们可恶意设计针对驱动基因的抑制剂,我们就有可能阻止肿瘤的进程。事实上,这也是我们现在看到的针对肿瘤的各色精准靶向疗法的核心支柱思路。 二、受体酪氨酸激酶里有大量癌症驱动基因,因此也富含经典抗癌靶点 引发生长失控的突变的驱动基因里,非常重要的一类,是酪氨酸激酶TK。TK是一类酪氨酸特异性蛋白激酶,以ATP为主的NTP可以作用于TK的酪氨酸残疾并使其磷酸化变构,从而实现细胞内的信号传导。酪氨酸激酶参与很多经典信号传导通路,包括各类生长因子对信号生长、增殖等信号的传递。此类激酶的过度持续激活,往往成为驱动癌变关键。 酪氨酸激酶又可以分为细胞膜上的受体酪氨酸激酶(Receptor Tyrosine Kinase, RTK)和细胞质和细胞核内的非受体酪氨酸激酶(non-Receptor Tyrosine Kinase, nRTK)。存在于胞内的nRTK中也有很多经典的抗癌靶点,比如2001年上市的伊马替尼(格列卫)靶向的ABL就是个胞内的非受体酪氨酸激酶(nRTK),诺华格列卫是全球首个重磅小分子精准靶向抗癌药,其颠覆性疗效深度改写了慢粒白血病的治疗格局。另外,JAK、BTK等也都是举足轻重的非受体酪氨酸激酶,相关的小分子抑制剂在肿瘤和自身免疫治疗中也起着重要作用。 不过,胞内信号传导蛋白有cross-talk的风险,位于细胞膜上的受体型酪氨酸激酶(RTK)原则上说,成药性可能会有优势。人体内目前已经鉴定58种RTK,主要分为几个亚类: 1. 表皮生长因子受体,Epidermal Growth Factor Receptor, EGFR家族;主要成员有EGFR/HER1/erbB1, HER2/erbB2, HER3/erbB3, HER4/erbB4; 2. 血小板衍生生长因子受体,Platelet Derived Growth Factor Receptor, PDGFR家族;主要包括PDGFRα、PDGFRβ、CSF-1R(击落刺激因子1受体)、干细胞因子受体c-kit; 3. 血管内皮生长因子受体,Vascular Endothelial Growth Factor Receptor, VEGFR家族;主要包括VEGFR1/FLT1, VEGFR2/FLK1, VEGFR3/FLT4;调节血管生成; 4. 成纤维细胞生长因子受体,Fibroblast Growth Factor Receptor, FGFR家族;主要包括FGFR1-4,可调节多种细胞生长、分化,在血管生成、伤口愈合、肿瘤发生等发挥重要作用; 5. 肝细胞生长因子受体,Hepatocyte Growth Factor Receptor, HGFR家族;是c-MET编码产物;肿瘤发生中重要作用,促进细胞增殖、抑制细胞凋亡、促进血管生成、促进肿瘤迁移及侵袭和转移等; 6. 胰岛素受体,Insulin Receptor, INSR家族;包括胰岛素受体、胰岛素样生长因子受体IGF1R等,在血细胞肿瘤当中常见此类受体高表达。
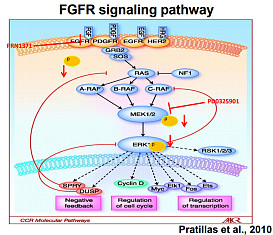
这里有非常多我们耳熟能详的经典靶点,以及针对这些靶点的经典药物: I. EGFR-TKI,靶向EGFR的酪氨酸激酶抑制剂(TKI)。2003年FDA批准阿斯利康的第一代药物吉非替尼(易瑞沙)上市,2015年FDA批准阿斯利康的第三代药物奥希替尼(泰瑞沙)上市。2011年药监局批准贝达药业的第一代药物埃克替尼在国内上市,2020年药监局批准豪森药业的第三代药物阿美替尼在国内上市。同时,国内有10多个三代EGFR-TKI新药在研,预计2020-2021年还会有多款国产1类新药获批上市。 II. HER2靶向药。1998年FDA批准基因泰克的曲妥珠单抗(赫赛汀)上市;2013年FDA批准基因泰克的靶向HER2的ADC药物TDMA(Kadcyla)上市。2018年药监局批准恒瑞医药的吡咯替尼国内上市,用于HER2阳性乳腺癌的治疗,同时吡咯替尼治疗HER2突变的肺癌治疗也在推进中。吡咯替尼在HER2+乳腺癌的2期临床中,展现出了比现在全球主流HER2小分子抑制剂更强的临床疗效,与金斯瑞的CAR-BCMA T、百济神州的BTK抑制剂一道,成为国内创新药由“me-too”向“me-better”攀升的标志。 III. 靶向多条RTK通路的多重激酶抑制剂。2005年FDA批准拜耳的索拉非尼(多吉美)上市,2015年FDA批准卫材的乐伐替尼上市。2014年药监局批准恒瑞的阿帕替尼国内上市,2018年药监局批准正大天晴的安罗替尼国内上市,2018年药监局批准和记黄埔的呋喹替尼国内上市。多重激酶抑制剂,往往同时抑制多条RTK通路,比如安罗替尼同时抑制VEGFR、PDGFR、FGFR、c-KIT等,索拉非尼同时抑制VEGFR、PDGFR、RAF、MEK、ERK等,相关通路对于肿瘤的发展往往有着重要的作用(比如血管生成),因此多重激酶抑制剂有着典型的广谱抗癌效果,比如3个获批的国产新药的首个适应症,阿帕替尼选了胃癌、安罗替尼选了非小细胞肺癌、和记黄埔选了结直肠癌;安罗替尼后续又批了软组织肉瘤、小细胞肺癌,甲状腺髓样瘤也在审批中;而索拉非尼在预后很差的肝癌、肾癌、甲状腺癌等适应症上仍然有着重要地位。对于那些目前治疗预后很差的实体瘤瘤种,多重激酶抑制剂多少都有一定效果,这为该类药物留出了非常多可拓展的空间。 三、FGFR小分子抑制剂开发现状与格局 在梳理了肿瘤驱动基因和受体型酪氨酸激酶(RTK)之后,FGFR靶点抑制剂在抗肿瘤中的作用就比较好理解了:作为RTK的重要分支,FGFR在肿瘤的发生发展过程中起着重要作用;在特定情况下,FGFR基因是肿瘤的驱动基因,成为潜在的抑制剂成药靶点。 FGFR全称是成纤维生长因子受体(Fibroblast Growth Factor Receptors),由胞外配体结构域和胞内酪氨酸激酶域构成的受体酪氨酸激酶,包括FGFR1\FGFR2\FGFR3\FGFR4四种亚型。正常条件下,FGFR参与包括胚胎发育、代谢平衡、组织修复和再生等生理过程;而一旦出现扩增、融合或激活型变异,将导致FGFR信号过度激活,进而促进细胞增殖、生存、肿瘤药物抵抗、血管生成及更重要的免疫逃逸。 和其它RTK一样,FGFR是位于细胞膜上的受体型靶点,其胞外区可以结合特定配体、从而实现信号的输入,其胞内区的酪氨酸激酶功能在结合配体后被激活、从而实现信号的输出,两个结构域的功能是协同进行的,继而实现相关胞外信号向胞内传递的过程。
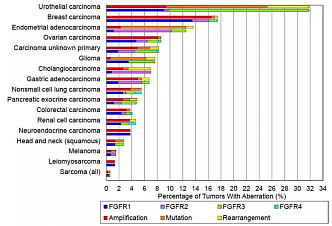
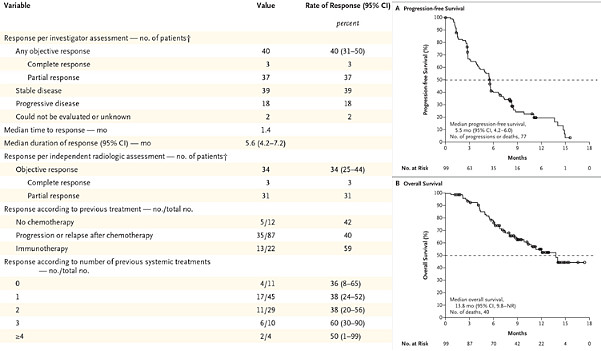
2016年一项涉及4853癌症患者的基因测序研究显示:7.1%的癌症患者,FGFR基因都有异常,最常见的是FGFR1基因扩增占50%。膀胱癌患者的FGFR异常比例最高,达到32%;乳腺癌为18%;子宫内膜癌13%,肺鳞癌13%,卵巢癌9%。当然,我们在第一段中分析过,癌症细胞中可能有大量的突变,不同突变的临床意义是不同的,不是说靶向抑制了突变基因就有抗肿瘤效果,而是需要靶向抑制了关键的驱动基因才能有显著的抗肿瘤效果。所以,虽然FGFR突变在不同肿瘤中广泛存在,并不等于FGFR抑制剂就有显著的广谱的抗肿瘤效果。 这里有一个前沿创新药都会面临的问题:很多概念听起来很好,但上了临床后的效果完全不及预期;背后关键是我们对细胞内相关生物学过程仍然知之甚少。所以,FGFR是不是、在哪些场景下是个好的抗肿瘤精准抑制靶点,仍然需要POC(Proof of Concept)概念验证。 过去10年中,国外科学家和相关药企已经开发了一系列FGFR抑制剂,并且做了一些列临床实践探索。在FGFR2突变或FGFR2/3融合的尿路上皮癌、FGFR2基因融合或重排的肝内胆管癌上,已经证实了突出的临床价值,并相应诞生了两款FDA批准上市的FGFR抑制剂新药(强生的Erdafitinib和Incyte的Pemigatinib)。另外,Fisogatinib在FGF19阳性的肝细胞癌的概念验证临床中,证实了FGFR4也是FGF19+HCC患者可靶向的驱动基因。 1. FGFR1-4抑制剂Erdafitinib已经获批尿路上皮癌治疗 Erdafitinib(Balversa)是强生与Astex合作开发的FGFR1-4选择性抑制剂。2019年4月,FDA批准erdafitinib二线治疗FGFR3突变或FGFR2/3融合的尿路上皮癌,获批基于II期临床。2019年7月NEJM(新英格兰杂志)发表了该II期临床的更新更全面数据:99患者参与,单药治疗客观缓解率ORR=40%,其中完全反应CR率=3%、部分反应PR率=37%,中位起效时间1.4月,中位缓解持续时间DOR=5.6月,无进展中位生存期PFS=5.5月,中位生存期OS=13.8月,12个月时总生存率为55%。13%患者因不良中止给药,无治疗相关死亡,主要不良包括FGFR抑制剂典型的高磷血症、低钠血症等。13%的不良中止给药,相比传统化疗药物,总体耐受性还算得上良好。 尿路上皮癌(Urothelial Carcinoma, UC)是膀胱癌的主要类型。传统二线是紫衫烷类或长春氟宁类单药治疗,客观缓解率ORR约为10%,中位生存期为7-9月;而一项免疫检查点抑制剂(PD1单抗)的研究,客观缓解率为13%-21%,中位生存期提高到10.3月。 可以看到,对于FGFR+UC患者而言,相比传统化疗“10%的缓解率 + 7-9月的生存期”和PD1单抗“13%-21%的缓解率 + 10.3月的生存期”,拥有“40%的缓解率+ 13.8月生存期” 的FGFR1-4抑制剂Erdafitinib是显著更好的治疗选项。Erdafitinib是膀胱癌的首个小分子靶向疗法;膀胱癌主要类型的晚期尿路上皮癌中,15%-20%的患者会有FGFR突变,对于这类患者,FGFR抑制剂有望改写治疗格局。 国内市场由强生开发。
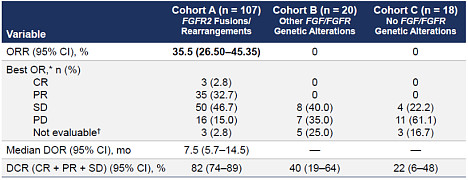
(原文:Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma,N Engl J Med 2019; 381:338-348) Erdafitinib在另一项187晚期实体瘤患者参与的I期临床中:有26名膀胱癌患者参与,客观缓解率也达到了46.2%。在该试验中还有其它实体瘤患者参与,包括有11名胆管癌患者,达到了27.3%的客观缓解率,并且实现了11.4月的中位缓解持续期,胆管癌也是值得重视的适应症。 2. FGFR1-3抑制剂Pemigatinib已经获批肝内胆管癌(iCCA) 近年随着二代测序NGS技术的兴起,iCCA已经鉴定出一些常见的突变,包括IDH1/2突变、FGFR2突变或融合,其中FGFR遗传畸变(主要是FGFR2融合)发生在10%-16%的iCCA患者中。FGFR2融合事件通常存在于非常高比例的肿瘤细胞中,最可能代表这是早期的原癌驱动突变,即FGFR2是FGFR2+iCCA患者的驱动基因,暗示了FGFR2抑制剂对于FGFR2+iCCA患者很可能有不错的效果。 Pemigatinib是选择性的FGFR1-3抑制剂,2020年4月获批二线治疗FGFR2融合或重排的肝内胆管癌(iCCA)。根据2020年3月在《The Lancet Oncology》(柳叶刀-肿瘤子刊),截至2019.3.22日,107例有FGFR2融合或重排的iCCA患者中,客观缓解率ORR=35.5%,其中完全缓解CR率=2.8%、部分缓解PR率=32.7%,疾病控制率DCR=82.2%。不过,该试验同时入组的其它FGFR遗传突变的B队列(20患者)中只有SD而没有CR或PR;而没有FGFR异常的C队列(18患者)中同样没有CR和PR且SD比例更低;暗示了FGFR抑制剂可能对FGFR非敏感突变的患者效果不佳(并不是所有类型的FGFR突变都对FGFR抑制剂有反应),而对于非FGFR的肿瘤患者可能基本没有作用。
肝内胆管癌是一种致命的原发性肝癌。原发性肝癌每年致死60万人,其中肝内胆管癌仅占原发性肝癌5%-10%,但过去30年里发病率从0.32人/10万人到0.85人/10万人,发病率不断增长。目前手术是唯一选择,接受保守姑息治疗的患者平均生存期仅1.8月,接受手术治疗患者平均生存12.2月,但手术治疗后复发患者的治疗选择非常有限。因此,FGFR抑制剂首次将肝胆管癌带入靶向治疗时代,有着非常显著的临床意义! 国内市场已经授权给信达生物开发。 3. 其它选择性FGFR抑制剂 Infigratinib(BGJ398)是BridgeBio制药的子公司QED开发的FGFR1-3抑制剂,在尿路上皮癌患者中有显著活性。根据2020年3月《Cancer》上发表的研究,区分了对上路尿路上皮癌UTUC和膀胱尿路上皮癌UCB的不同活性:在8例UTUC入组的队列中,1例CR+3例PR,客观缓解率ORR达到50%,其余均实现了SD,使得疾病控制率DCR达到100%;而在UCB队列中,客观缓解率ORR=22%,疾病控制率DCR=59.3%。之前公司在2018 ESMO公布的二线治疗FGFR2+iCCA的2期结果:71名患者入组,客观缓解率ORR=25.4%,疾病控制率DCR=83.6%,中位缓解持续时间DOR=5.4月,中位无进展生存期PFS=6.8月,中位生存期OS=12.5月。另外在BGJ398治疗实体瘤的132人入组的I期临床中:33名晚期尿路上皮癌患者,ORR=36%,包括1名CR患者肿瘤完全消失;21名FGFR扩增的肺鳞癌患者,疾病控制率DCR=58.9%。 Fisogatinib(BLU-554)是Blueprint开发的选择性的FGFR4抑制剂,据2019年10月《Cacner Discovery》公布的1期数据:在肝细胞癌HCC患者中,FGF19阳性患者客观缓解率ORR=17%(FGF19是FGFR4的配体),而FGF19阴性患者客观缓解率ORR=0%,证实了FGFR4是FGF19+HCC可靶向的驱动基因(targetable driver)。2018年全球HCC新发84.1万、死亡78.1万,一线获批包括多激酶抑制剂索拉非尼和乐伐替尼等,另外有多种激酶抑制剂和PD1单抗作为二线治疗,中位PFS和中位OS分别保持3-7个月和9-13个月,并不理想。肝癌高度异质性,临床治疗方案一直进展艰难,有大量未被满足的需求。临床数据表明大约30%的HCC患者存在FGF19/FGFR4信号通路的异常激活,暗示了fisogatinib成为HCC靶向药物的潜力。BLU-554国内已经授权基石药业在做临床开发。 Derazantinib(ARQ 087)是Basilea开发的选择性的FGFR1-3抑制剂,在做针对FGFR2融合的iCCA适应症二线治疗的1-2期临床。其中29人入组的可评估队列中,总体客观缓解率ORR=20.7%,疾病控制率DCR=82.8%,估计中位无进展生存期PFS=5.7月,中位OS尚未达到,但估计95%下限是13.4月;27.6%患者出现三级或以上不良反应。在FGFR2+iCCA适应症中,表现出了与Pemigatinib相仿的强劲疗效潜力。国内已授权给仑胜医药开发。 AZD4547是阿斯利康开发的选择性的FGFR1-3抑制剂,对FGFR1有很强的亲和力。之前公布的15名FGFR1扩增的鳞状细胞肺癌患者,13名可评估患者中1人实现部分缓解、ORR=8%,2名患者无进展时间达到12周、对应中位无进展生存期4.9个月,这个效果不算太突出。检测的FGFR1扩增情况与疗效不太线性相关,说明了肿瘤的强异质性。2019年11月和誉生物获得AZD4547的全球独家开发权。和誉生物旗下自有FGFR4靶向药物ABSK011在研,另有全球Best-in-Class潜力的高特异性CSF-1R小分子抗肿瘤抑制剂ABSK021进入临床,并引进了X4的Firs-in-Class的口服CXCR4拮抗剂的大中华区开发权。 TAS120是大鹏药品和大冢制药共同开发的FGFR1-4抑制剂。在1期试验中,28例FGFR2融合iCCA患者,客观缓解率ORR=25%,疾病控制率DCR=79%,保持了FGFR抑制剂在FGFR2+iCCA中的良好表现。值得注意的是,在3名接受BGJ398和1名接受Debio1347(另一种FGFR抑制剂)治疗后耐药进展的患者中,TAS120单药治疗使得2名患者获得了PR、2名获得了SD,缓解持续时间5.1月-17.2月,而疾病控制率DCR=100%!在FGFR抑制剂耐药后,用其它FGFR抑制剂仍然实现了100%的DCR,这为FGFR的序贯治疗以强化疗效,提供了潜在的巨大的延展空间。 ASP5878是一款高选择性的FGFR1-4抑制剂。在一项纳入FGFR通路异常的晚期尿路上皮癌、肝癌、肺鳞癌的I期研究中,整体疾病控制率DCR接近40%,其中肝癌DCR=41.7%,尿路上皮癌DCR=38%,总体中位无进展生存期PFS=2.67月,中位进展时间TTP=2.76月。 LY2874455也是一款口服的选择性的FGFR1-4抑制剂。根据2017年发表的数据:在24可评估患者的剂量爬坡队列中,1人PR、14人SD,客观缓解率ORR=4.2%、疾病控制率DCR=62.5%;在12名可评估患者的非小细胞肺癌扩展队列,有11人SD,疾病控制率DCR=91.7%;在15可评估患者的胃癌拓展队列,有1人PR、12人SD,客观缓解率ORR=6.7%、疾病控制率DCR=80%。疾病控制率不错,但客观缓解率ORR表现一般。 除了上述产品之外,国外在研FGFR抑制剂还有CH5183284、PRN1371等。国内在研的FGFR抑制剂还有:诺诚健华的ICP-192是泛FGFR抑制剂,针对胃癌、胆管癌等,ICP-105是FGFR4抑制剂,针对肝癌;海和生物的德立替尼靶向FGFR1-3和其它激酶靶点,在做2-3期;亿腾景昂的小分子泛FGFR抑制剂EOC317在做1期;珍宝制药由药明康德开发的泛FGFR抑制剂HZB1006在做1期;贝达药业的BPI-17509,针对胆管癌,在做1期;滴露对和海和生物的FGFR抑制剂HH185在做1期等。 除了选择性的FGFR抑制剂,还有不少的多重靶点激酶抑制剂也兼具FGFR抑制功能,包括:2012年FDA批准上市的Ponatinib(靶向Abl-PDGFR、VEGFR2、FGFR1、Src等)、2014年FDA批准上市的Nintedanib、在临床后期的Dovitinib、MK2461、ODM203等等,包括正大天晴获批的安罗替尼也可以同时靶向VEGFR\PDGFR\FGFR\c-kit等。不过,一体两面地看,多重靶点带来多能性的同时、也会牺牲精准性。对于那些FGFR是明确驱动基因的应用场景,高选择性的FGFR抑制剂仍然是更好的选择。 四、小结: 基于肿瘤驱动基因的抑制剂靶向药开发正在迅猛发展——生物学发展,尤其是二代测序带来的遗传信息的爆炸式发展,极大促进了基于肿瘤驱动基因的小分子靶向抑制剂(相当比例是酪氨酸激酶抑制剂)的发展;FGFR是个值得充分重视和探索的赛道与靶点——受体酪氨酸激酶里有大量癌症驱动基因,因此也富含经典抗癌靶点;FGFR是其中重要一支,对其生物学作用的理解和药物开发在过去10多年中有了长足进步,正在改写肝内胆管癌和尿路上皮癌的治疗格局,仍然有大量潜在价值有待进一步挖掘。 $信达生物(01801)$ $诺诚健华-B(09969)$ 注:文中提到的标的仅供逻辑展示,不构成推荐或者持仓暗示。 |
【本文地址】