实验篇 |
您所在的位置:网站首页 › FAT基因家族 › 实验篇 |
实验篇
|
实验篇——基因家族成员鉴定(一) 文章目录 前言一、TBtools介绍1.1 TBtools能做什么1.2 主界面介绍 二、基因家族成员鉴定2.1 获取序列文件2.2 提取出CDS序列2.3 将CDS序列翻译为AA序列2.4 准备参考序列2.5 第一次BLAST对比2.6 删除重复项2.7 得到第一次比对后的AA序列2.8 第二轮BLAST比对2.9 根据保守结构域筛选2.10 (额外)推断Pr的功能和结构 三、后续问题补充3.1 没有注释文件 总结 前言今天认识了生信分析中一个十分实用的软件——TBtools ,我将带大家初识一下该软件,并使用该软件进行基因家族成员鉴定。 有关生物知识,请看 生物笔记——注解(一) 一、TBtools介绍系统学习推荐: https://www.yuque.com/cjchen/hirv8i 1.1 TBtools能做什么 Sequence Toolkits, 序列处理与操作BLAST, 序列比对与可视化GO & KEGG, 基因集合功能分析Graphics, 生物信息学数据可视化Others, 暂未划分功能About, TBtools相关信息与运行设置 1.2 主界面介绍
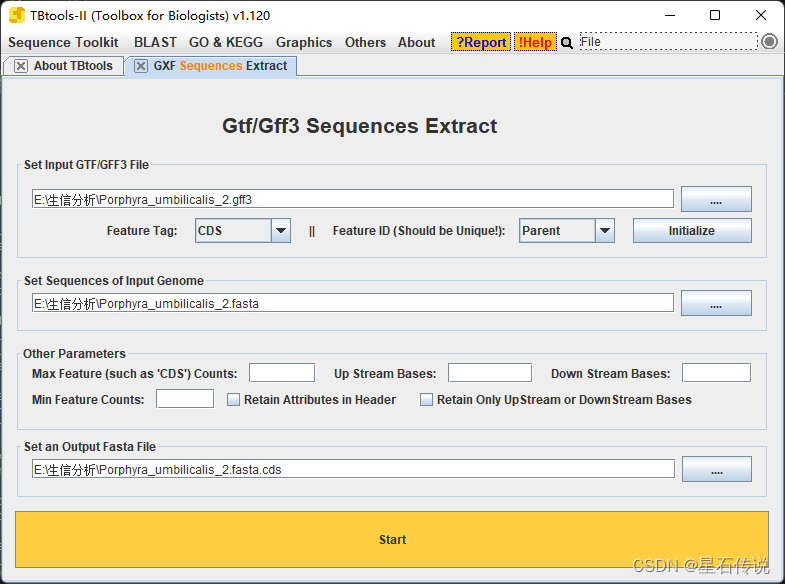
一. Fasta Tools Fasta Extract (Recommended) —序列提取/截取 Fasta Stat —返回Fasta 序列文件的详细信息, Fasta ID Simplify ----简化 Fasta 序列 ID Fasta Rename ----对序列ID进行重命名 Fasta ID Prefix —进行一定的ID调整,加前缀。 Fasta to Table Convert —转换成 ID 和序列在同一行的表格模式 Merge and Split —进行Fasta序列文件合并或者分割 Sequence Pattern Locate —知道某些序列模式在给定的序列集合出现的具体位置和频次 SSR miner —基于输入的 Fasta 序列文件,快速挖掘并鉴定可用的 SSR 位点。 Fasta Window Stat —对 Fasta 序列进行滑窗统计,比如对基因组序列滑窗统计 GC含量分布,N含量等 二. NCBI Sequence Fetch GenBank to Fasta —将 GenBank 格式转化为Fasta 格式 NCBI Sequence Download (Basic) —一个简单的 NCBI 序列下载功能 Bulk NCBI Sequence Download (Advanced) —这一功能用于批量下载 NCBI 序列 由此可见,该软件中包含了许多程序,方便在图形化界面中操作,而后续功能不再赘述,当用到时再详细介绍,接下来主要介绍基因家族成员鉴定流程。 二、基因家族成员鉴定推荐视频: https://www.bilibili.com/video/BV16i4y1P7zq/?spm_id_from=333.999.0.0&vd_source=e34f9443a7e5ae37b9a8faac238fc132 基因家族成员鉴定是指通过比对和聚类分析来识别具有相似序列或功能的基因。 基因家族是指在基因组中存在多个拥有相似序列或功能的基因。 这些基因通常由基因复制、基因重组或基因转座等机制产生,形成一个基因家族。 2.1 获取序列文件待分析的基因组序列文件的下载可来自各种数据库,在这以NCBI数据库为例。 可直接从NCBI官网下载,在搜索栏输入Accession号,打开对应的页面。在页面上找到"Send to"(发送给)下拉菜单,然后选择"File"(文件)。或者在TBtools软件中下载,打开Bulk NCBI Sequence Download (Advanced)程序。但是第一种方法只能一个一个的下载(当要下载极多序列文件时,费时间)。 而第二种方法则是能批量下载,但是它会全部下载到一个文件里。(我们要的是分开的序列文件)。 以上两者方法不够快速,试着用代码实现: import os from Bio import Entrez # 用于与NCBI进行通信 from Bio import SeqIO # 用于处理序列数据 def wht_jiyinduqu(EMAIL, Accession, output_folder, geshi, mingzi): """ EMAIL:邮箱地址 Accession:待下载序列的标识号 output_folder:保存序列的文件夹 geshi:下载序列的格式 mingzi:文件名,与Accession一一对应 """ # 设置您的邮箱地址(用于NCBI数据访问政策) Entrez.email = EMAIL # 定义要下载的序列的Accession号列表 with open(Accession, "r", encoding="utf-8-sig") as f: accessions = f.read().split() # 创建保存序列的文件夹 os.makedirs(output_folder, exist_ok=True) with open(mingzi, "r", encoding="utf-8-sig") as f: MZ = f.read().split() # 检查accessions和filenames列表长度是否相等 if len(accessions) != len(MZ): print("错误:两个文本文件行数不一致,请检查") exit() for i in range(len(accessions)): accession = accessions[i] filename = MZ[i] + f".{geshi}" filename = os.path.join(output_folder, filename) # 创建文件名 handle = Entrez.efetch(db="nucleotide", id=accession, rettype=geshi, retmode="text") # 以FASTA格式返回一个文本文件,其中包含下载的序列数据 record = SeqIO.read(handle, geshi) # 将获取的fasta格式序列转化为序列记录 handle.close() # 关闭与NCBI的连接 SeqIO.write(record, filename, geshi) # 将当前序列记录以fasta格式写入指定的文件 print(f"下载完成: {filename}") #多线程形式执行多个程序 import threading # 创建线程对象 thread1 = threading.Thread(target=wht_jiyinduqu("您的邮箱", "Accession号.txt", "Se1", "fasta", "mingzi.txt")) thread2 = threading.Thread(target=wht_jiyinduqu("您的邮箱", "Accession号.txt", "Se2", "gb", "mingzi.txt")) # 启动线程 thread1.start() thread2.start() # 等待线程结束 thread1.join() thread2.join()果然,还是代码快,比如我要分别下载24个Accessions基因序列对应的fasta和gb格式的文件。若是一个一个下载,几乎要100分钟,而我爬取数据最多不超过十分钟。近乎十倍的效率差,且只需要机器自动爬取。(代码若有待优化之处,请指出,谢谢) 2.2 提取出CDS序列 进入GXF Sequences Extract 程序导入gff3文件,点击”Initialize"选择"Feature Tag" 为 CDS,“Feature ID”为Parent设置物种的基因组序列,选择fasta文件“Other Parameters” 定制提取的具体内容设置输出文件路径,为cds文件。最后点击"Start“。
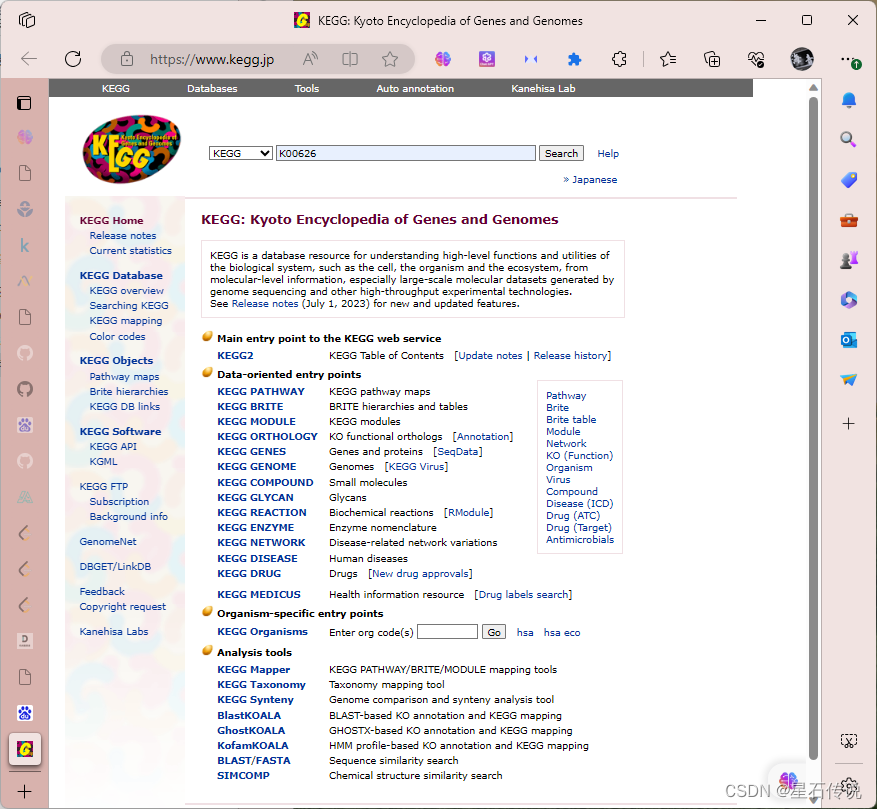
一旦确定了基因家族,你可以在公共数据库(如KEGG)中搜索该基因家族的参考序列。下载AA序列 使用基因家族的名称、基因名或相关关键词来进行搜索,如使用酶分类号K00626在打开的界面中搜寻其他物种(如拟南芥ATH)的对应的基因为参考序列• 在大多数网页浏览器,按下键盘上(Ctrl + F),将会打开一个浏览器的内置搜索框。 在打开的搜索框中,输入您要查找的基因的关键词,比如基因名称、基因ID等。
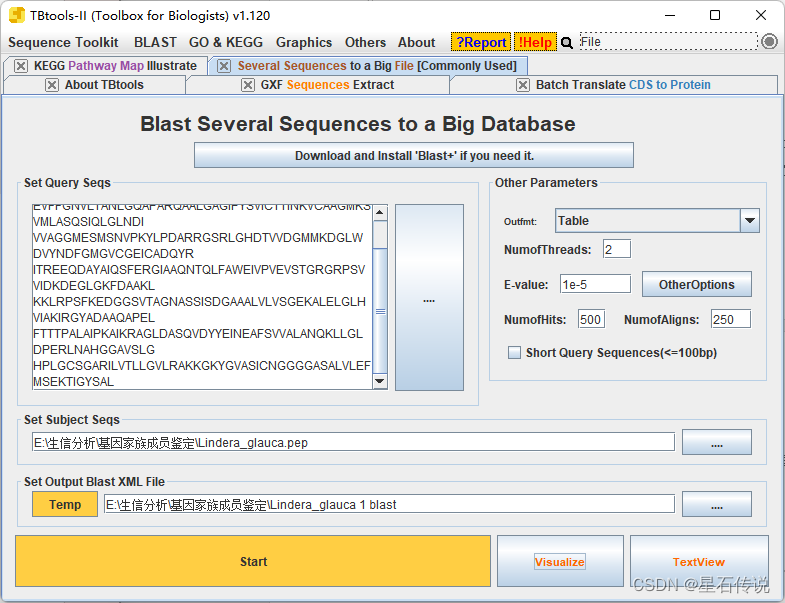
进入 ”Several Sequences to a Big File [Commonly Used]"程序 注意: 您输入的待鉴定的AA序列文件应该为.pep格式
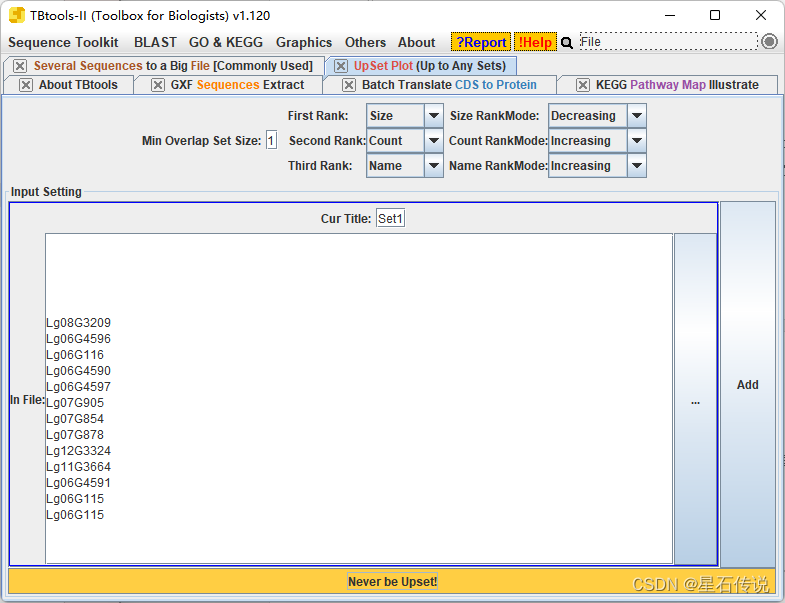
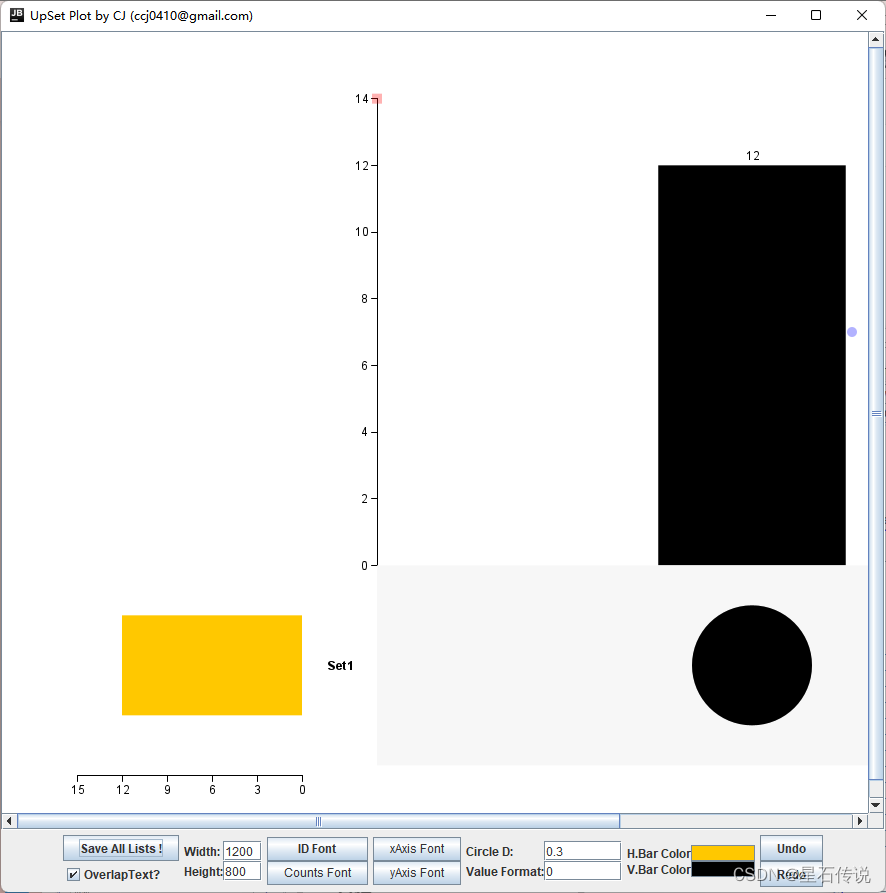
进入"UpSet Plot (Up to Any Sets)“程序 将上一步得到的blast比对后得到的文件的第二列复制进来 点击"Never be Upset!”,进入图形 最后,双击图形中的柱形,会弹出删去重复项的基因ID
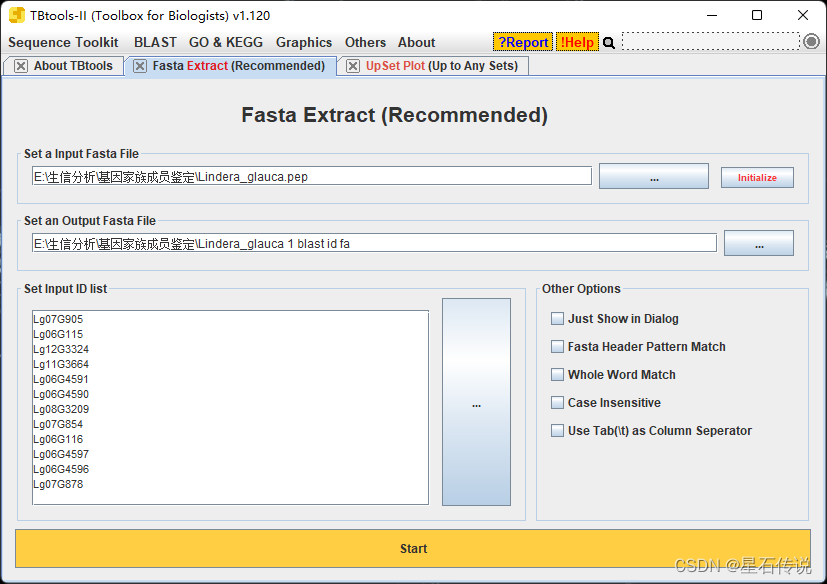
将上一步得到的ID复制进“Fasta Extract (Rcommended)"程序
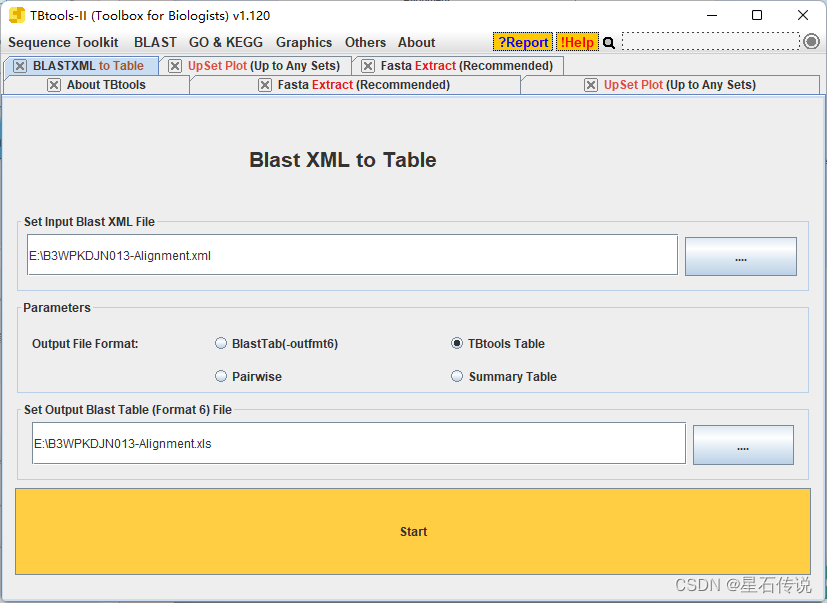 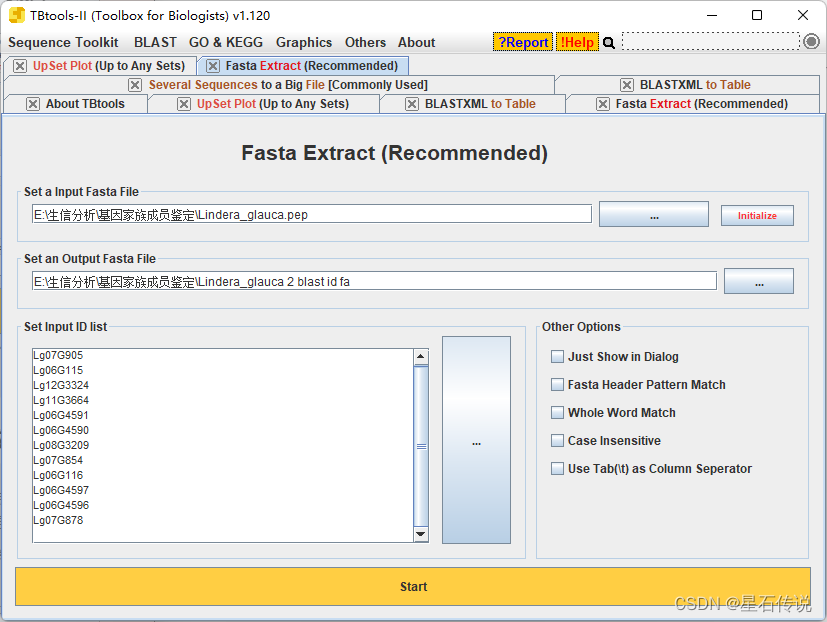 2.9 根据保守结构域筛选
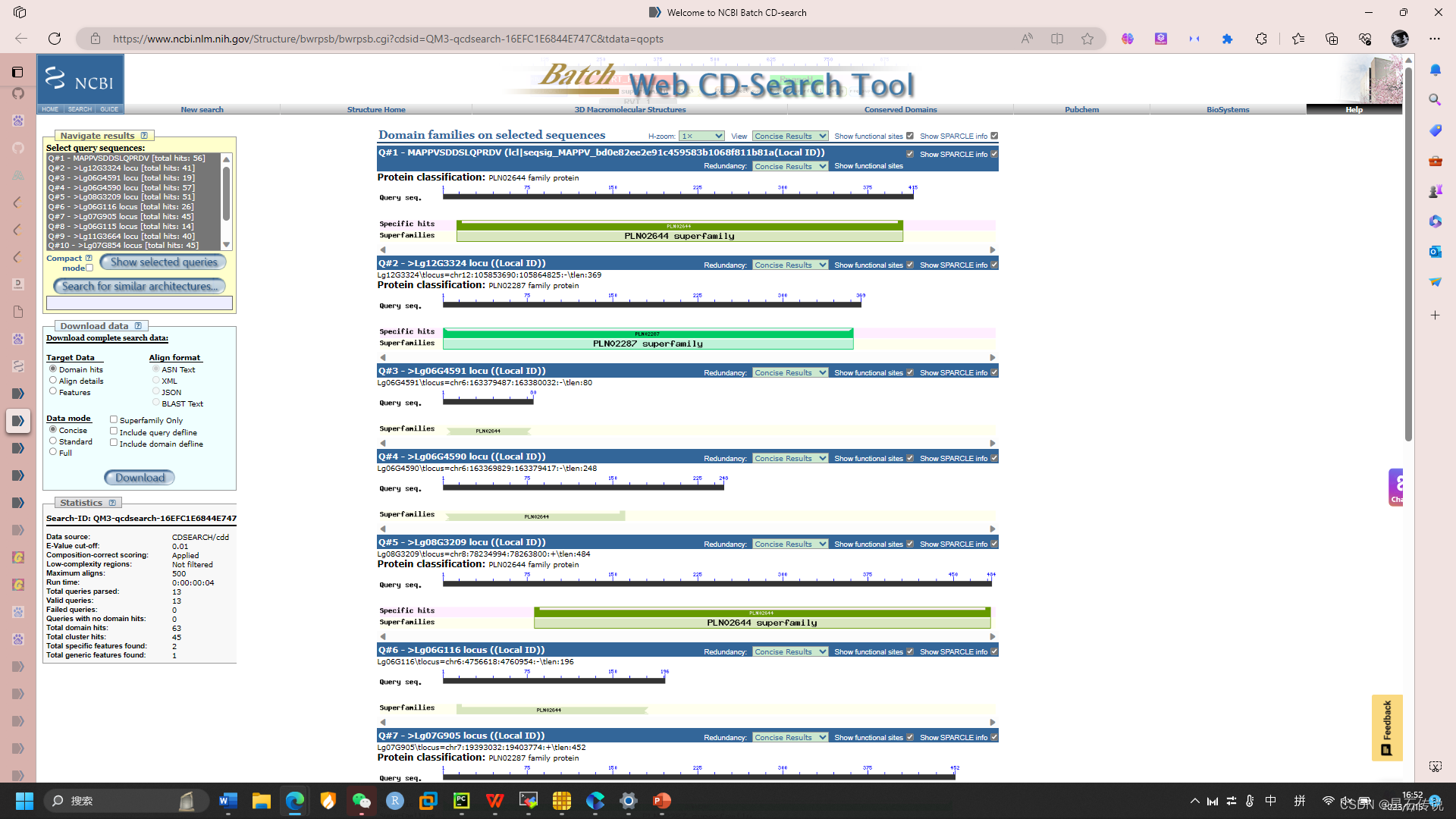
进入NCBI官网,点击"Proteins",再点击"Conserved Domain Database(CDD),再点击"Batch CD-Search”进入页面后,导入经第二轮blast的文件(可以将参考序列导入,以便观察),其它默认不变,点击“Submit”进入页面后,点击:Browse results",全选“select query sequences:”框,再点击"show selected queries"查看有无与参考序列相似结构域,若无,则舍弃该基因。最后整理出有相似结构域的基因AA序列的ID名称,归为参考序列的基因家族。
2.9 根据保守结构域筛选
进入NCBI官网,点击"Proteins",再点击"Conserved Domain Database(CDD),再点击"Batch CD-Search”进入页面后,导入经第二轮blast的文件(可以将参考序列导入,以便观察),其它默认不变,点击“Submit”进入页面后,点击:Browse results",全选“select query sequences:”框,再点击"show selected queries"查看有无与参考序列相似结构域,若无,则舍弃该基因。最后整理出有相似结构域的基因AA序列的ID名称,归为参考序列的基因家族。
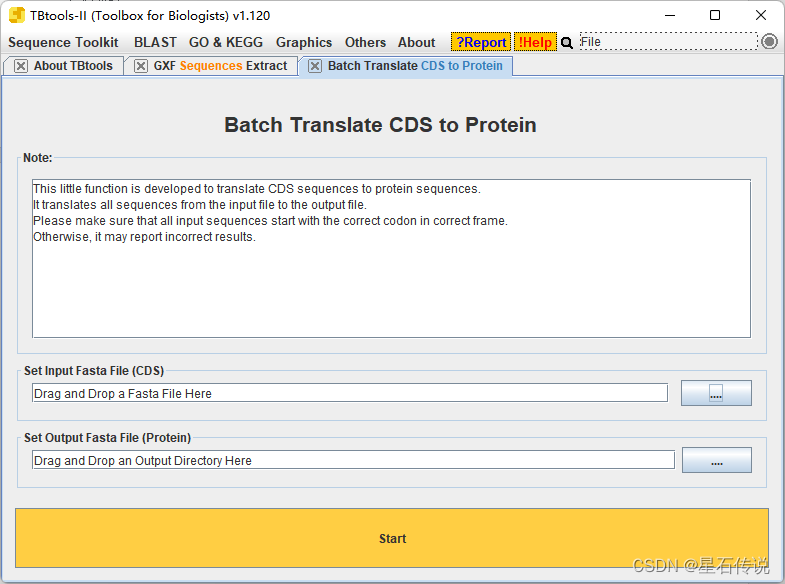
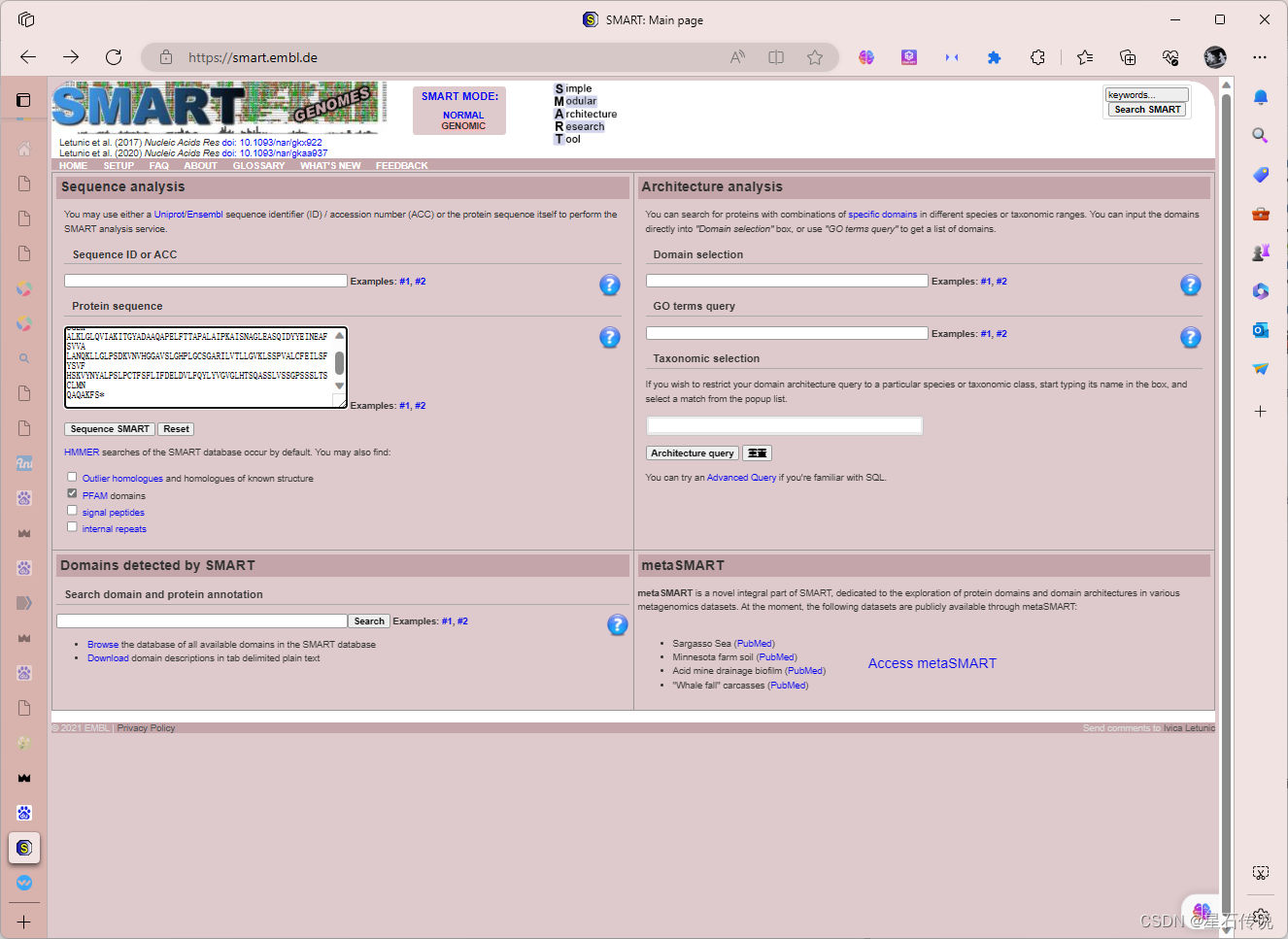
选取不同参考序列,将待进行基因家族成员鉴定的基因的AA序列,与其分别进行以上步骤,最后完成每个基因的鉴定 2.10 (额外)推断Pr的功能和结构1.进入SMART数据库 2. 将AA序列一个一个的放入"Protein sequences“框中,勾选"PFAM domains”,再点击"Sequence SMART“。 要是物种只有基因组的 .fasta文件,没有注释文件,这种情况如何做呢? 怎么说呢,如果实在没有,那就只能通过预测来得到CDS序列。 在TBtools软件中 进入”Complete ORF Prediction (Batch)”程序,输入你的fasta格式的基因组文件,设置输出的cds文件的路径。 点击“start” 它会得到 3个文件: CDS 文件蛋白序列文件无法预测到有效 ORF 的序列 ID 信息。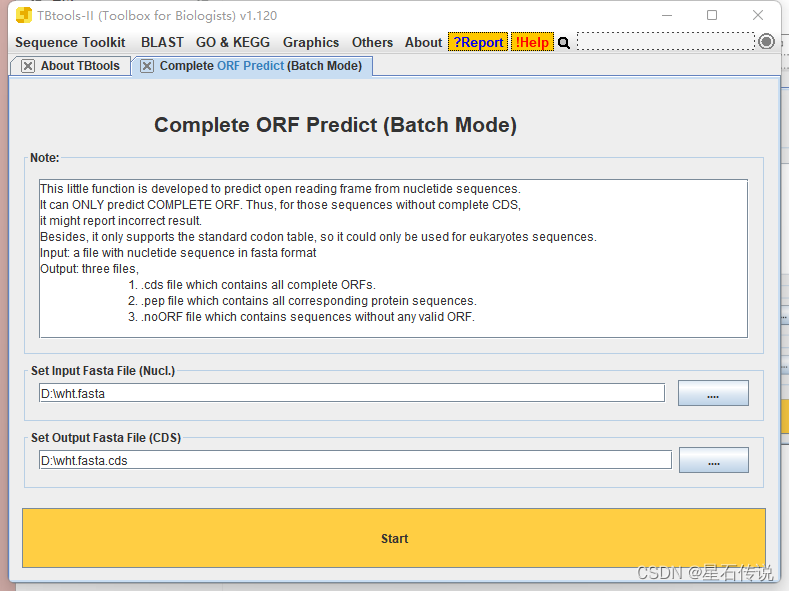 这样就通过预测得到了我们所需的CDS文件,甚至直接一并得到了AA序列文件,这样就可以继续进行后面的步骤。 还是那句话,这样预测得到的,无法保证准确性,如果有注释文件还是更好。
总结 这样就通过预测得到了我们所需的CDS文件,甚至直接一并得到了AA序列文件,这样就可以继续进行后面的步骤。 还是那句话,这样预测得到的,无法保证准确性,如果有注释文件还是更好。
总结
基因家族成员鉴定的主要流程: 获取物种基因组的fasta文件和gff3文件获取参考序列经过两轮BLAST比对保守结构域筛选,鉴定基因家族换参考序列,反复进行3,4步骤,直到完成鉴定这篇文章是我学习基因家族成员鉴定的笔记,亦是我的第一篇有关生信分析的文章,或许还有许多不够详细之处,请理解。 孤帆远影碧空尽,唯见长江天际流。 -2023-7-14 实验篇 |
【本文地址】
今日新闻 |
推荐新闻 |