生信分析系列 |
您所在的位置:网站首页 › 8001wj冠 › 生信分析系列 |
生信分析系列
|
数据处理 获得干净的序列数据后,接下来就可以做比对分析了。比对分析是指把各条序列与参考序列相同位置上的碱基作比较,以确定突变信息。 比对工具有很多种,小编用的比对工具是MAFFTv7.450,使用方法可以参考官方文档(https://mafft.cbrc.jp/alignment/software/)。目前公认的参考序列的GISAID编号是EPI_ISL_402125。同时,GISAID上也有比对好的结果文件供下载。 2 分析方向经过以上的预处理后,就可以开始进行我们的数据挖掘工作了。分析的方向有很多,这里简要地介绍一下突变分析和进化分析。 突变分析 通过比对我们知道了各个突变所处的位置,要进一步统计这些突变发生在哪个区域,需要对参考序列进行注释。注释是指确定病毒序列上的编码区,例如最重要的编码病毒S蛋白在序列中的位置。 序列的注释信息可以从NCBI中获得(https://www.ncbi.nlm.nih.gov/sars-cov-2/)。通过统计突变出现的次数,我们可以知道哪些是高频突变以及它们出现的编码区,并作进一步的深入分析,如图2是小编分析的各种高频突变在新冠基因组上的分布。
比如,之前受到很大关注的突变D614G(对应图2中的A23403G突变)出现在编码新冠病毒S蛋白的区域中,而S蛋白是与细胞接触进而感染细胞的关键。为了解这个突变与病毒的传染能力是否有关,我们可以进一步分析这个突变是否改变了S蛋白的结构导致病毒与细胞的亲和力更高等。另外,对共同突变的分析可以揭示新冠病毒的多样性。 例如,小编发现根据共同突变能够把病毒分为18个病毒亚群,它们的分布在各大洲有明显的区别(图3)。
进化分析 通过进化分析,我们可以追踪病毒的演变情况,推断病毒的传播路径以及估计病毒出现的时间等。 BEAST是对病毒做进化分析强有力的工具,被应用在很多包括新冠在内的病毒研究中。BEAST实际上是一组工具套件,其中包括BEAUti,BEAST和TreeAnnotator。BEAUti的作用是生成一个参数文件,BEAST读取这个参数文件后进行分析得到结果,最后TreeAnnotator对BEAST产生的结果作进一步的处理。接下来小编跟大家分享一下如何使用这款工具。 首先准备输入文件。BEAUti接受Nexus和Fasta格式的序列文件,如果输入文件不是这两种格式,则需要进行转换,这里小编使用Aliview(https://ormbunkar.se/aliview/)进行格式转换。接下来要设置一系列的参数,这里小编给出自己在分析过程中使用的参数(表1),对参数更加详细的解释请参考官方文档(https://beast.community/workshop_rates_and_dates)。
得到的参数文件输入到BEAST中然后执行程序,完成后会产生两个文件,一个是log文件(以.log结尾),另一个是trees文件(以.trees结尾)。Trees文件需要用TreeAnnotator作进一步处理,这里需要设置BurnIn参数,如果选择Burnin(as states),则一般设置为马尔科夫链步数的10%;如果选择Burnin(as trees),则一般设置为树总数的10%(步数与树总数的数量关系为1000:1)。产生的树文件小编推荐使用iTOL(https://itol.embl.de/)进行可视化。图4是小编分析的结果,并且把上文提到的18个病毒群使用iTOL进行了颜色标注。
Log文件需要用Tracer(https://beast.community/tracer)进行读取,这里burnin会默认设置为马尔科夫链步数的10%,可以手动调整。在Traces栏目中,会显示一系列的统计结果,当统计结果的ESS大于200时,这个结果可信度更高,如果ESS |
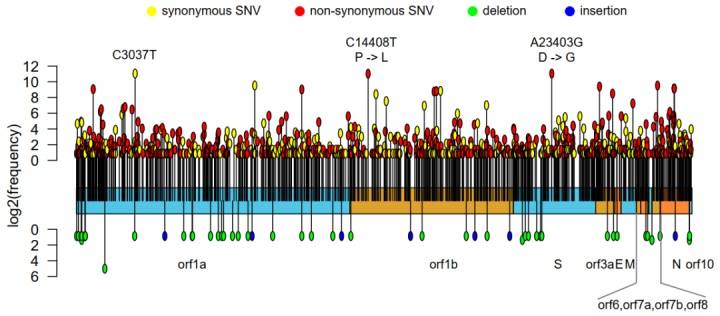 图2
图2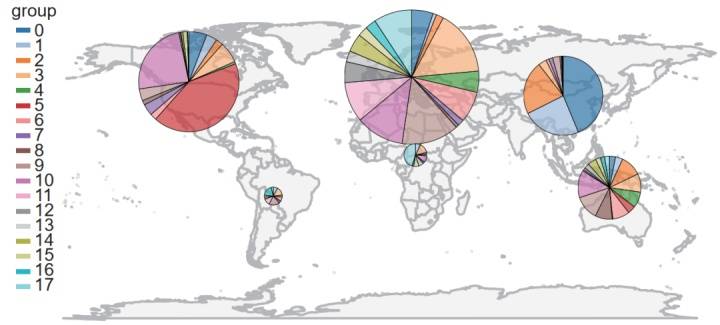 图3
图3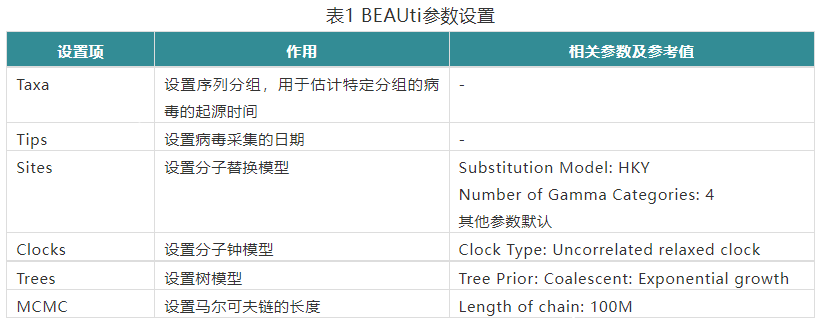
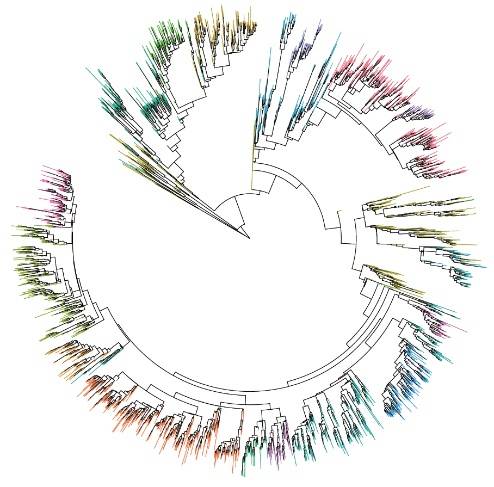 图4
图4【本文地址】