通过505(b)(2)途径批准的药物综述:揭示药物发展趋势和监管要求 |
您所在的位置:网站首页 › 2类新药临床试验要求有哪些 › 通过505(b)(2)途径批准的药物综述:揭示药物发展趋势和监管要求 |
通过505(b)(2)途径批准的药物综述:揭示药物发展趋势和监管要求
|
申报者常常面临这样的挑战:研究是否能支持505(b)(2)途径的批准?这项为期5年(2012-2016年)的回顾性分析对FDA网站上已批准的505(b)(2)项NDAs进行了综述,以确定研究的性质(临床前、临床药理学和有效性/安全性),并更好地了解监管要求的趋势。 该数据库有226个NDAs,其中112个有完整的FDA审查信息。最普遍的类型有: 类型3,新剂型; 类型4,新组合; 类型5:新处方或新制造商。因为它们可以显示针对不同类型的申请的趋势对,所以我们对这112个NDA进行了进一步的研究。基于对NDA审查文件的调查,结合指导性文件,推导出需要进行研究的决策树,可以为成功实施505(b)(2)开发计划和提交NDA提供建议指南。 — 关键词 — NDA,临床药理学,缓释,速释,固定剂量联合用药 — 背景 — 1999年,FDA发布了涵盖505(b)(2)章节的工业应用指南草案,介绍了联邦食品、药物和化妆品法案部分,并描述了某些监管方面,如资格、提交和专利/独占保护。根据定义, 505 (b)(2)申请是“一份包含安全性和有效性调查的完整报告的申请,但申请批准所用的信息至少部分来自其他研究者的数据”。505(b)(2)是一种新药申报途径,包括新剂型、新给药途径和新适应症。 通过505(b)(2) 提交的药物可以用其他研究者研究的数据获得批准,这要依赖于(1)对之前已批准的药物的安全性和有效性(AFSE)的研究; 和/或(2)其他研究者已发表的临床和临床前研究文献数据。这不仅需要通过对相对生物利用度(BA)或生物等效性(BE)研究成功地桥接到RLD(参比制剂),而且可能也需要一些潜在的额外研究,以充分支持新产品的有效性和/或安全性。如果申报方能够得到原始数据(比如可以参考的权利),那么可以按NDA的补充申请或505(b)(1)路径申报。 鉴于近来人们对505(b)(2)途径的兴趣日益浓厚,制药行业需要这方面的临床开发方案的具体指导。除了505(b)(2)的具体指导草案外,FDA还发布了关于“是否决定提交ANDA或505(b)(2)申请”的指导草案,指导申报者进行申报(仿制药 vs 505(b)(2)), 有一篇文章对批准505(b)(2)的NDA的临床药理学方面进行了综述。 然而,这些指导原则并不能提供指导药物研发的细节。由于505(b)(2)申请被视为NDA,因此,美国联邦法规21章(21CFR)21标题第1章(如第320.27、320.24、320.25、320.22、300.50、314.50部分)和各种FDA指导性文件提供了提交NDAs所需的设计和需进行研究的一般信息。关键在于理解FDA的期望,为了避免505(b)(2)路径下新产品的数据对支持审批的缺陷,需要和审评机构进行pre-IND或其他FDA会议。 表1 提交FDA的类型
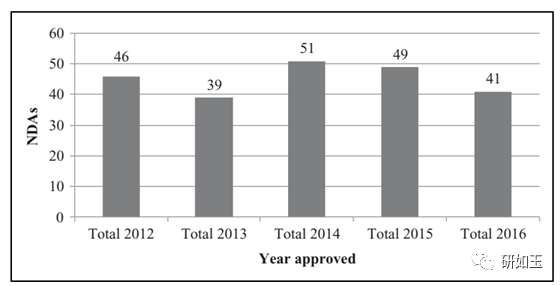
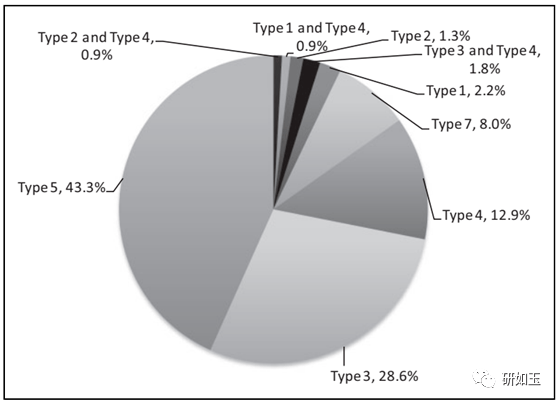
简称: NDA,新药申 a:一种含有2种或2种以上活性成分的新药组合。 本回顾性分析不仅提供最近批准的505(b)(2) NDA(2012-2016)的概述,同时总结了支持NDA批准的研究趋势。重要的是,在没有全面了解监管历史的情况下,对产品的临床研究可能已经满足(1) FDA的要求,(2)其他机构的要求,(3)申请者的选择。 然而,根据FDA提交的分类类型(表1)所定义的新药相对于RLD所做的特定类型的修改,本文将讨论有关临床前、临床、特别是临床药理学研究的建议,以支持505(b)(2) NDA。这篇综述的最终目标是给505(b)(2)的申请者向药物开发组织提交文件提供指引。 材料和方法 在本回顾性分析的第一部分中,对FDA网站进行了搜索,检索了2012年1月至2016年12月间所有通过505(b)(2)的NDA,信息来自于“NDA和BLA批准年历”。另外,对过去的5年关于505(b)(2)的信息,如提交FDA分类类型、剂型、给药途径,从FDA的drug@FDA数据库里收集,还从FDA的橙皮书数据库里获得所需的补充信息。 在第二部分,对所选产品的药物审批包进行了深入的回顾,重点是这些NDA审查部分:摘要、医学、药理学和临床药理学生物药剂学。只包含审查资料可获得的品种。我们建立了一个数据库,包括RLD、临床药理学研究的类型、临床前和临床/疗效或安全性研究的存在与否,以及FDA在临床前(pre-IND)或批准前(pre-NDA)交流会议上的建议。 在我们研究和最后讨论中只保留可以确定研究类型趋势的NDAs。这篇综述重点在于这些研究背后的性质和基本原理,并提供了决策树,该决策树是基于对各种通过505(b)(2)途径批准的口服药物产品的趋势和对各种FDA指导文件的深入总结而得出。 结论和讨论 从2012年到2016年批准的505(b)(2)NDAs的总结 在这5年内,共有226个505(b)(2)的NDAs产品被批准。每年具体情况见图1。 在这226个产品中,FDA提供了224个产品的分类类型,见图2。可以看出,最多的是类型5(新处方或新制造商,43.3%),类型3(新剂型,28.6%),紧跟的是类型4(新复方,12.9%)。新的化学实体和新的活性成分(类型1和类型2)很少见,因为这些品种大部分是按505(b)(1)NDAs提交的。这种分类能让研究者筛选和更好的理解通过505(b)(2)途径申报药品的性质。然而,即使是FDA已经明确的类型,也会有各种差异(比如类型3,就包括改变处方、新的给药途径、更换辅料或新的剂量)。
图1. 从2012年到2016年FDA批准的505(b)(2) 对提交505(b)(2)产品的主流类型的研究 在前面说的226个NDAs品种中,181个可以在FDA网站中查到药物批准包。另外,对于112个主流类型(类型3、4、5)品种的分析足以看出申报者申报类型的趋势。实际上,有51个NDAs被排除,因为他们属于少数的类型(图2),是非典型的505(b)(2) (n=5),而且作为类型7提交的18种NDAs也没有包括在该文中,因为他们和RLD相比并没有变化。
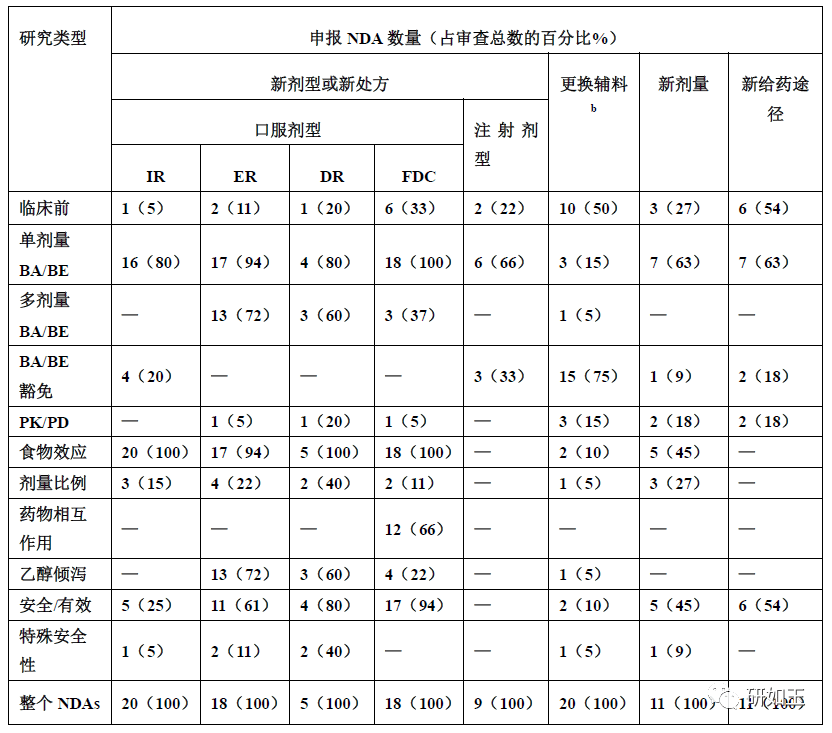
图2. 2021年到2016年提交FDA的505(b)(2)的类型(n=224)。类型说明见表1。 表2列出505项(b)(2)中类型3、5和4(分别为新剂型、新处方或新组合)所需进行的研究。 表2 505(b)(2)不同类型需要进行的研究a
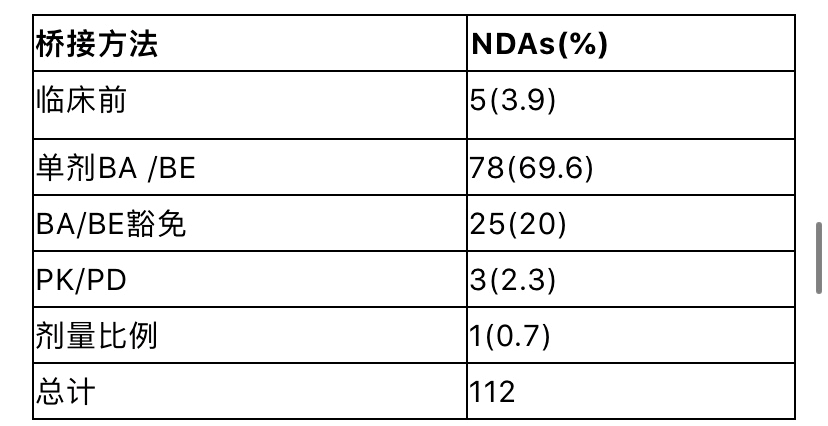
a 本表列出了根据药品的改变而纳入特定研究的NDAs的数量。 b 含量改变、增加或移除辅料 IR:速释,ER:缓释,DR:迟释,FDC:固定剂量组合 在对NDAs的回顾中很少发现以下研究:肝肾损伤患者的药代动力学研究、QT延长研究、药物间相互作用评估对酶、转运体抑制/诱导的影响,或联合用药、临床前药理学、遗传毒性、致癌性、生殖毒性研究。因为FDA已经要求参比制剂解决了这些缺陷,所以这些研究没有列入表2。因此,接下来主要讨论常做的研究。 参比制剂(RLD)的选择 如果申报者需要已批准药物的安全有效(AFSE)信息来申报,那对相关的参比制剂进行科学的桥接是必须的。选择RLD必须基于API一致。对说明书的逐项比较可以确定最合适的RLD(例如给药途径、处方),同时要记住,申报品种和RLD之间的相似程度可能会对要进行的研究数量产生影响。 然而,在开始新药研发之前,FDA相关部门应该同意参比制剂的选择。在某些情况下,当选定的RLD不再可用(例如,由于安全和有效性以外的原因而停止使用),可以从通过ANDA途径批准,并在Orange Book中指定为RS的产品中选择。然而在这种情况下,安全性和有效性数据来自于停止使用的原始的NDA产品。在某些开发产品的过程中,可能会使用多个RLDs(例如,针对固定剂量组合[FDC]产品,申报者将两种已获批准的口服药物组合在一起)。 桥接方法 69.6%的NDAs(n=112)通过单剂量BA/BE的研究比较新药和RLD的药代动力学来进行科学桥接(表3),这符合FDA工业指南的涵盖关于505(b)(2)部分的规定。即使做了相对生物利用度(BA)研究,BE并不是必须得证明的。实际上,产品可能会出现吸收速率和/或BA程度(例如Cmax和/或药时曲线下面积AUC)同RLD有差异,原因在于处方的变化(比如新的长效制剂),或是预期改善产品PK性质(比如让药物更快起效)。 如表3所示,其他的科学桥接方法也偶尔被采用。例如,PK/PD研究可以作为支持一种新的药物释放系统批准的桥梁方法(例如,RLD为速释制剂[IR],新品种为一种新的控释制剂[MR]),与RLD相比,新品种可能在PK方面存在一些差异。为了证明BA的差异不会导致不同的治疗结果,需要对暴露量——药效关系有一个很好的理解。 表3 桥接方法
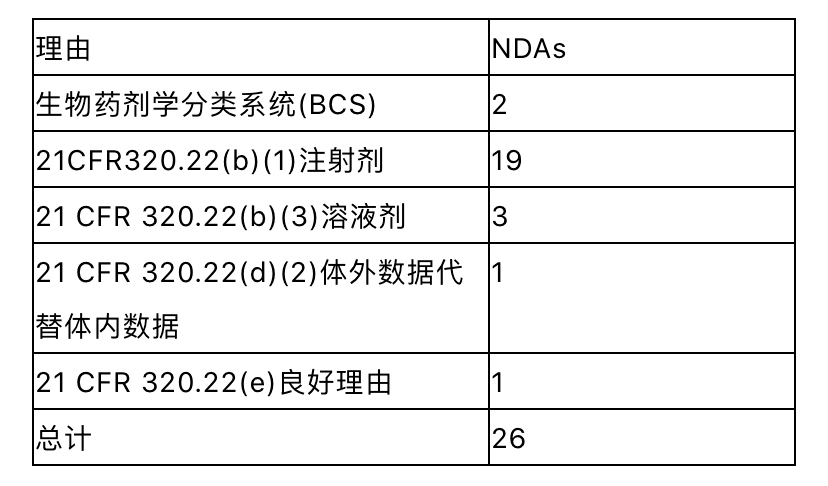
然而,有20%的NDAs,申报者参考了RLD,并依赖于AFSE,但没有进行任何与RLD相关的衔接研究。表4显示了支持体内BA/BE豁免的理由。 表4体内BA/BE豁免的理由
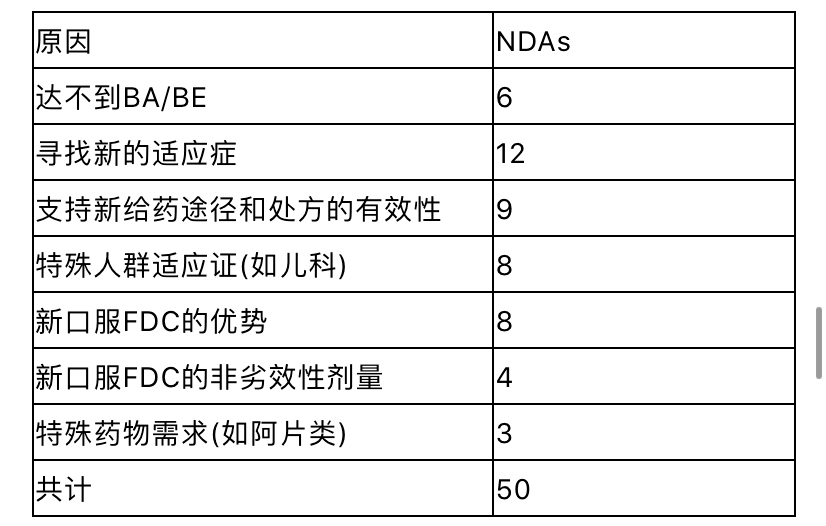
BA/BE豁免建立在两个主要基础上。首先,根据FDA对行业的指导原则“基于生物药剂学分类体系的即时释放固体口服制剂豁免体内生物利用度和生物等效性研究”,只要制剂中使用的辅料不显著影响主药的吸收,BCS I类(高溶解-高渗透)和BCS III类(高溶解-低渗透)药物可不需要进行体内BA/BE试验。 其次,根据 21CFR 320.22(b),“体内生物利用度或生物等效性的豁免标准”的条款,药品的BA/BE可以自证豁免。在文中谈及的NDAs,这些标准应用于静脉注射用溶液制剂(n=19)和口服液(n=3)。然而在这些情况下,审评机构建议申报人提供足够的理由说明拟议产品和RLD之间的任何差异(如成分及组合物、适应症、稳定性、配制说明、pH值和渗透压、递送体积、输注速率)对临床安全性和有效性的潜在影响, 例如NDA207131头孢唑啉钠静脉注射产品。 食物效应研究 对于几乎所有505(b)(2)口服给药产品,无论其如何变化(表2),需要评估食品对新药BA的影响。在一些速释(IR)和调释(MR)NDAs案例中,已经进行了空腹和餐后条件下的BE研究(试验品vs RLD),而这些试验通常用于ANDAs产品提交。然而根据“食物影响生物利用度和餐后生物等效性研究的行业指南”,对于NDAs包括505(b)(2),通常建议进行空腹条件下的BA/BE研究(试验品vs RLD)和新产品的食物效应研究(空腹 vs 餐后)。 基于我们的经验,可以有某种方式简化开发:将空腹BA/BE研究与食物效应相结合进行评估,通过一个三交叉试验,也就是试验品和RLD在空腹条件下,与一个额外的第三臂试验品餐后条件下试验,从而比较空腹和餐后。药监部门通常接受这种实验方案,它可以节省时间和减少开发费用。 临床有效性/安全性研究 在112个NDAs中有50个示例,申报者不能仅仅依靠RLD的AFSE,除了进行BA/BE研究之外还进行了临床疗效/安全性研究。 然而,即使满足BA/BE的标准,对于表5中所列的情况,也要做临床有效性/安全性研究。 表5 需要进行临床疗效/安全性研究的原因
就新的固定剂量组合药品(FDC)而言,应按照21CFR 300.50(a)中的规定,证明FDC相对于每种单一治疗均有优势。这些信息可以从公开出版的文献或从早先的NDAs获得。例如已经在欧洲获得批准的口服精氨酸培哚普利和氨氯地平的FDC产品,NDA 205003, FDA认为参考文献并不证明该组合产品优于最高剂量的各个单方。因此我们进行了一项III期临床试验,比较了最高剂量下联合用药和单方用药,以证明具有临床优势。 此外,当FDC产品的剂量范围与每个单一RLD不同时(例如一天一次vs 一天两次)非劣效剂量桥接需要满足法规的要求,要证明所依赖的RLDs数据是适当的。例如卡格列净/二甲双胍的FDC产品(NDA 204353),申报人被要求证明通过使用稳健的模型和模拟策略,或者通过比较两种给药方案的临床疗效,每天给卡格列净一次的疗效与之前推荐的每天给药两次相比,疗效不会发生变化。在这种特殊情况下,主办方选择进行临床试验。 有时还偶尔观察到其他类型的安全性研究,例如评估防止药品滥用(即人药品滥用可能性的研究);新局部外用药品的皮肤安全性研究(即评估致敏性、光敏性、长期刺激性、防晒系数);口腔崩解片在咽部的安全性评价(NDA 208025, 204326)。 临床前研究 在某些情况下,申报者需要做些额外的临床前研究(例如安全性、致癌性/基因毒、生殖毒,和/或药物联用的评价),比如新剂型或新给药途径(n=11)、更换辅料(n=8)、新的口服FDC产品(n=6)。表6总结了需要做的临床前研究,在API、辅料、给药途径和拟联合用药变化后,这些临床前研究用于支持缺乏足够的文献信息或人体研究数据的品种申报。 表6 API、辅料、给药途径需考虑的其他非临床研究
ADME:吸收、分布、代谢、排泄;AFSE:管理机构先前的安全性/有效性调查; API:活性药物成分;FDC:剂量固定组合物;PD:药效动力学;PK:药代动力学;RLD:参比制剂ADME:吸收、分布、代谢、排泄;AFSE:管理机构先前的安全性/有效性调查; 例如6个新给药途径的NDAs产品(3个注射、1个眼用、1个局部外用、1个透皮给药),RLD是口服给药,申报人要做临床前的药理学/毒理学桥接研究来支持新临床途径的安全性。 而且在新处方的情况下,申报者不仅仅评估了新加辅料目前的安全性,还评估了辅料对API暴露量的影响。以NDA 208090(一种新的羟考酮缓释胶囊)为例,需要进行额外的临床前研究(ADME研究、长期毒性、生殖毒性和致癌性研究),以解决PK数据的缺乏,并评估API在用新辅料后的临床暴露情况。 最后,对于口服固定复方产品(FDC),每个单方的安全性数据要充分,要能支撑组方使用:合适的适应症、剂量、治疗时长(如慢性或急性用药)、目标人群。然而,由于新的组方缺乏人体前的安全性试验,所以必须进行评价,特别是两个API可能影响药效活性和毒性的情况。例如,NDA206439(盐酸美金刚缓释制剂+盐酸多奈哌齐),申报人要做额外的临床前试验去评估毒性、剂量范围和对中枢神经系统的可能的协同效应。 多剂量BA/BE和体内乙醇剂量倾泻 多剂量的PK研究要评估药物达到稳态时,API吸收进入体循环的速率和程度。一般来说,单剂量的PK研究被认为是一种更灵敏的BA/BE研究办法。因此包括505(b)(2)在内的大部分BA/BE的桥接研究都是单剂量研究。不过,有20个NDAs除了单剂量BA/BE研究外,还做了多剂量BA/BE。这些NDA包括新的缓控制剂(MR)和迟释制剂(DR)。 FDA的指导原则“提交NDAs或INDs的BA和BE研究之一般考虑”描述了什么时候需要进行多剂量BA/BE研究,包含有缓释制剂的例子。实际上当给药剂量不同时,为了可靠地比较相对PK的暴露程度,经常需要用缓释制剂达到稳态时的相对PK暴露量(例如,超过24小时的AUC)和速释制剂(或作为参比制剂的缓控释制剂)稳态时的PK对比。 对于迟释制剂(DR),一些FDA指导原则提出要证明BA/BE和速释制剂相同,这意味着要进行空腹条件下的单剂量药品BA / BE的研究以及食物效果研究。不过,对于我们表中的5个NDA迟释制剂而言,申办者还进行了多剂量BA / BE研究。这背后的理由尚不清楚,可能归因于迟释制剂复杂的释放特征。 而且,为了证实缓释制剂的效果,申请者应该做体外试验以排除酒精引起的缓释制剂剂量倾泻的可能(即缓释制剂在短时间内迅速释放药物,可能危及患者的安全)。如果体外试验结果表明有计量倾泻,审查要求申报人做服用酒精情况下的体内BE研究。 剂量比例研究 剂量比例研究评估给药剂量的增加是否与暴露量(AUC或Cmax)成比例地增加。FDA的工业指导原则“提交NDAs或INDs的生物利用度和生物等效性研究的一般性考虑”说明具有剂量范围内非线性PK特征药物的缓释制剂,应将缓释制剂的最高剂量和最低剂量下单剂量的PK与对应的参比速释制剂进行比较。 此外,当新药的剂量在组成比例上不相似时(即活性/非活性成分的比例不同),应进行单剂量空腹等效性评估研究或剂量比例研究。申报者对15个NDAs进行了剂量比例研究,请求批准这些新制剂。因此,他们或是进行剂量比例研究,或进行每种剂量比较各自RLD的BA/BE。 然而,其他的新剂量的NDAs申请,基于充分的体外溶出试验数据,申请人要求豁免低剂量的NDAs,或者根据21 CFR第320.22节,体内BA/BE是不证自明的。 药物—药物相互作用研究(DDI) 18个NDAs中的12个固定剂量组合产品(FDC),药物相互作用的PK研究评估两个药物可能的相互影响。尽管没有口服固定剂量组合产品的FDA指南,NDA的审查还是清楚地表明:如果之前没有进行过DDI研究,并且不能排除相互作用的可能性,则应在FDC的治疗组成之间进行DDI研究。开展这样一项研究也是遵循“固定剂量组合、共包装药品和先前批准的单方治疗艾滋病的抗逆转录病毒药物”的FDA工业指导原则的建议。 新口服制剂和新的口服固定剂量组合物(FDCs)的研究趋势 所有的临床前、临床药理。临床有效性/安全性相关的法规和科学要求都应该在提交NDA的申请中被满足。本文对2012-2016年批准的NDAs的全面总结揭示了对RLD改动最频繁的研究类型(即新处方和新口服FDC)的趋势。 新口服制剂 在这种情况下,申请者申请了505(b)(2)途径,以将药品从速释(IR)剂型(RLD)改变为另一种,或从改变为缓释(ER)制剂。在临床药理学研究方面,88%的NDAs进行了单剂量BA/BE研究和食物效应研究。然而,对于新的缓释制剂,还进行了多剂量BA/ BE研究和体外酒精剂量倾销研究(72%)。18%的NDA进行了剂量比例研究,34%的NDA进行了临床疗效/安全性研究。 新口服固定组合物 在这种情况下,两个RLDs的剂型可能是一致的(都是IR/IR,或MR/MR),或是不一致(IR/MR)。 取决于口服固定组合物(FDC)的剂型(IR/MR),无论是新的速释制剂(IR)或新的缓释制剂(ER),只要是“新的口服剂型”均需要临床药理研究。这两种情况下,应该对FDC进行BA/BE交叉研究,和同剂量的单一的APIs制剂做对比。 总的来说,在18个NDAs中,有17个口服FDCs做了临床疗效/安全性研究,以支持拟申请的适应症。 决策树 申请人如果想申报505(b)(2),应就其发展计划预先专门征询相关监管机构的意见。然而,这里仍然可以提出一些一般性的建议。基于对505(b)(2)申请审查中观察到的趋势,以及对各种相关FDA指南的研究,我们提出了决策树,用于帮助主要的505(b)(2)类型确定临床研究类型(图3-6)。
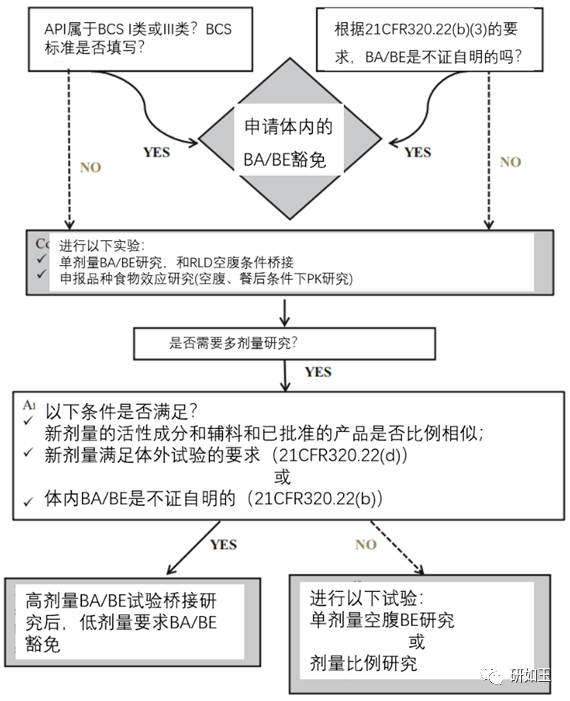
图3. 新速释制剂(IR)的临床药理决策树 * 关于生物豁免,即使某些申报者的产品符合生物豁免的条件,一些申报者仍可能希望进行至少一项BA / BE研究(例如,出于全球营销考虑)。
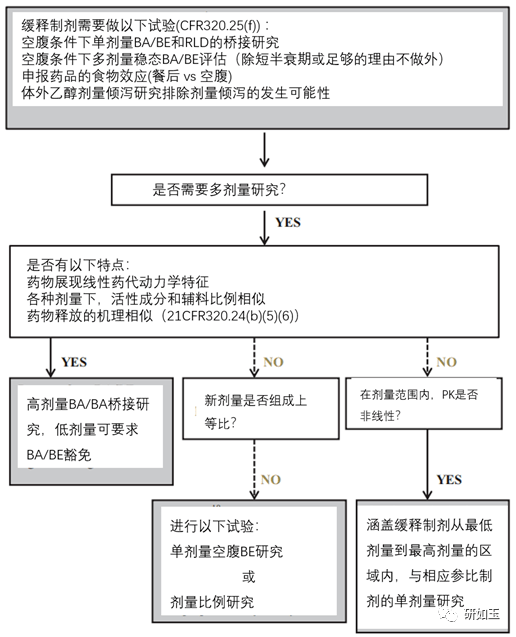
图4. 新缓释制剂(ER)的临床药理决策树
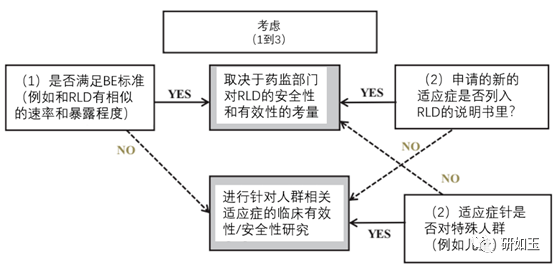
图5.口服制剂有变化的情况下临床疗效/安全性决策树。 * 新药品所需充分和良好控制的临床III期疗效研究的数量依赖于和RLD的药时曲线的相似性。如果对研究药物的PK / PD关系有充分的了解,若被研究药物的PK/PD的关系被很好理解得话,则PK / PD研究可能会代替临床疗效研究
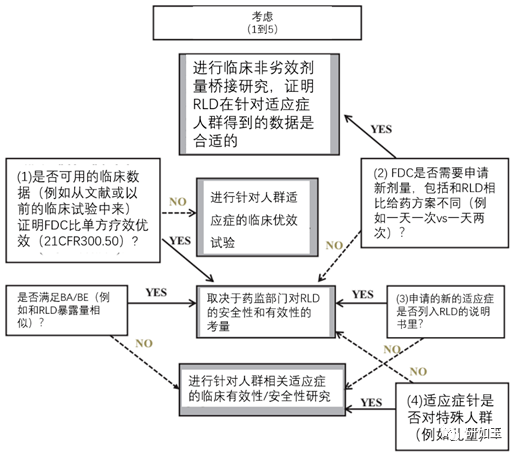
图6. 新的口服固定剂量组合物(FDC)的临床疗效/安全性决策树。 * 新药品所需充分和良好控制的临床III期疗效研究的数量依赖于和RLD的药时曲线的相似性。 结论 505(b)(2)已成为医药工业发展的主流。我们对2012-2016年批准的NDAs的全面审查揭示了505(b)(2)主要类型的研究趋势。 虽然确实没有关于505(b)(2)药物开发的具体指南,但大多数涉及NDA的FDA指南文件都不仅有助于对所进行的研究有合理的理解,而且还可以指出需要做什么才能通过505(b)(2)途径获得批准。该文构建的决策树可以为与FDA审评部门的会议上提出和确认开发策略提供依据。 作者注释 本文已于2017年11月13日在加利福尼亚州圣地亚哥的美国药物科学家协会(AAPS)年度会议上发表。 转载声明:本文转载自「研如玉」返回搜狐,查看更多 |

【本文地址】
今日新闻 |
推荐新闻 |